
- |<
- <
- 1
- >
- >|
-
Hee-Seon Park, Chang-Seob Seo, Charith UB Wijerathne, Hye-Yun Jeong, O ...2019 年42 巻1 号 p. 1-9
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/11/01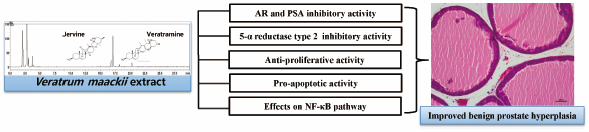 ジャーナル フリー HTML
ジャーナル フリー HTMLVeratrum maackii (VM), a perennial plant in the Melanthiaceae family, has anti-hypertensive, anti-cholinergic, anti-asthmatic, anti-tussive, anti-fungal, anti-melanogenesis, and anti-tumor activities. Here, we investigated the therapeutic effect of VM on benign prostatic hyperplasia (BPH) in human normal prostate cell line (WPMY-1) and a testosterone propionate-induced BPH animal model. WPMY-1 cells were treated with VM (1–10 µg/mL) and testosterone propionate (100 nM). BPH in rats was generated via daily subcutaneous injections of testosterone propionate (3 mg/kg) dissolved in corn oil, for 4 weeks. VM (150 mg/kg) was administered daily for 4 weeks by oral gavage concurrently with the testosterone propionate. All rats were sacrificed and the prostates were dissected, weighed, and subjected to histological, immunohistochemical, and biochemical examinations. Immunoblotting experiments indicated that WPMY-1 cells treated testosterone propionate had increased expression of prostate specific antigen (PSA) and androgen receptor (AR), and treatment with VM or finasteride blocked this effect. In rat model, VM significantly reduced prostate weight, prostatic hyperplasia, prostatic levels of dihydrotestosterone (DHT), and expression of proliferation markers such as proliferating cell nuclear antigen (PCNA) and cyclin D1, but increased the expression of pro-apoptotic Bcl-2-associated X protein (Bax) and the cleavage of caspase-3. VM administration also suppressed the testosterone propionate-induced activation of nuclear factor-kappaB (NF-κB). Our results indicate that VM effectively represses the development of testosterone propionate-induced BPH, suggesting it may be a useful treatment agent for BPH.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (4609K) HTML形式で全画面表示 -
Shudong Yang, Haiying Bao, Hui Wang, Qingjie Li2019 年42 巻1 号 p. 10-17
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLInonotus hispidus is an anti-tumour drug used in folk medicine. (4S,5S)-4-Hydroxy-3,5-dimethoxycyclohex-2-enone (HDE) is a compound isolated from Inonotus hispidus for the first time. However, the mechanisms underlying its therapeutic effects have not been elucidated. In this study, the in vitro screening, in vivo anti-tumour effects, mechanism of action, pharmacokinetics, and tissue distribution of HDE were investigated. HDE could inhibit the proliferation of HepG2 cells. Additionally, its half-maximal inhibitory concentration was 7.9 µg/mL. Increasing HDE concentrations significantly increased apoptosis rate in a dose-dependent manner. Furthermore, HDE was rapidly absorbed into mouse plasma, reaching a maximum concentration at 30 min. The area under the plasma HDE concentration–time curves for the studied organs were as follows: spleen > liver > lung > kidney > muscle > thymus > heart > brain. HDE also inhibited tumour growth up to 69%. The weights of organs harvested from HDE-treated mice were not significantly different from those harvested from control mice. Furthermore, HDE upregulated Fas expression and downregulated FasL expression in HepG2 cells. HDE significantly increased caspase-3 and caspase-8 activity. The anti-tumour effect of HDE might be realized by activating the Fas-mediated apoptotic pathway. We also found that HDE undergoes enterohepatic circulation or is quickly absorbed by the body, and the drug is released back into systemic circulation. In conclusion, HDE significantly inhibited H22 hepatocarcinoma cells (H22)tumour growth in mice without damaging organs; therefore, it may be suitable for treating liver cancer.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (4285K) HTML形式で全画面表示 -
Hipólita Lagunas-Herrera, Jaime Tortoriello, Maribel Herrera-Ruiz, Gab ...2019 年42 巻1 号 p. 18-25
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLHypertension is a disease of high prevalence and morbidity where vascular inflammation and associated oxidative stress (endothelial dysfunction) is the underlying cause of this pathology. We are reporting the antihypertensive activity of extracts and fractions of Malva parviflora in mice with chronic and acute hypertension. Also, the treatments of this plant were able to counteract the kidney inflammation and associated oxidative stress. The chronic hypertension model consisted of administration of angiotensin II (AGII) during 12 weeks, causing a sustained increase in systolic (SBP) or diastolic (DBP) pressure, with values of pharmacological constants of: ED50 = 0.038 mg/kg y Emax = 135 mmHg for SBP and ED50 = 0.046 mg/kg y Emax = 98 mmHg for DBP. The chronic hypertension caused the inflammation and lipid peroxidation in kidneys, measured by of tissue level of cytokines such as interleukin-1β (IL-1β), IL-6, Tumor Necrosis Factor-α (TNF-α), IL-10 and malondialdehyde, and treatments for M. parviflora were able to modulate these parameters. The chemical fractionation allowed to identify three compounds: oleanolic acid, tiliroside and scopoletin, which were tested in a model of acute hypertension. The pharmacodynamic parameters for SBP were ED50 = 0.01 and 0.12 mg/kg while Emax = 33.22 and 37.74 mmHg for scopoletin and tiliroside, respectively; whereas that for DBP data were ED50 = 0.01 and 0.02 mg/kg; with an Emax = 7.00 and 6.24 mmHg, in the same order. M. parviflora, is able to counteract the effect of chronic and acute administration of AGII, on hypertension, but also the inflammatory and oxidative damage in the kidney. The oleanolic acid, scopoletin and tiliroside are the compounds responsible for such activities.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (728K) HTML形式で全画面表示 -
Riho Tanigaki, Riku Takahashi, Mai Thanh Thi Nguyen, Nhan Trung Nguyen ...2019 年42 巻1 号 p. 26-33
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Tumor necrosis factor α (TNF-α), a pro-inflammatory cytokine, regulates inflammatory and immune responses by up-regulating gene expression in a manner that is dependent on the transcription factor nuclear factor κB (NF-κB). In the present study, we found that 4-hydroxypanduratin A and isopanduratin A, constituents of the rhizomes of Boesenbergia pandurata, inhibited the TNF-α-stimulated up-regulation of intercellular adhesion molecule-1 (ICAM-1) in human lung adenocarcinoma A549 cells. 4-Hydroxypanduratin A and isopanduratin A also reduced ICAM-1 mRNA expression and NF-κB-responsive luciferase activity in TNF-α-stimulated A549 cells. Moreover, 4-hydroxypanduratin A and isopanduratin A prevented the TNF-α-stimulated translocation of the NF-κB subunit p65 to the nucleus and the phosphorylation and proteasomal degradation of the inhibitor of the NF-κB α protein. The present results revealed that 4-hydroxypanduratin A and isopanduratin A inhibit TNF-α-stimulated gene expression and the NF-κB-dependent signaling pathway in A549 cells.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (6789K) HTML形式で全画面表示 -
Liqiang Qiu, Changwu Xu, Hong Jiang, Wenjing Li, Suiyang Tong, Hao Xia2019 年42 巻1 号 p. 34-42
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/11/03 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) and the chronic inflammation regulated by various inflammatory factors are the major pathological processes in the development of neointimal hyperplasia and in-stent restenosis after angioplasty. Cantharidin is a potent and selective inhibitor of protein phosphatase 2A, which plays pivotal roles in cell cycle progression, cell fate, and inflammation. This study was to explore whether Cantharidin could inhibit VSMCs proliferation, migration and inflammation. Transwell migration assay, Cell Counting Kit 8 and flow cytometry were performed. Western blot, Quantitative real-time PCR, and enzyme-linked immunosorbent assay (ELISA) were used to detect the expression of the markers. Results showed that Cantharidin remarkably suppressed VSMCs proliferation and migration induced by platelet derived growth factor (PDGF)-BB. Meanwhile, Cantharidin could significantly inhibit the phosphorylation of Akt (P-AKT) and p38 mitogen-activated protein kinase (MAPK) (P-p38), the expression of p38 MAPK (p38), and also the phosphorylation level of nuclear factor-kappaB (NF-κB) p65 (p65). Cantharidin obviously inhibited the expression of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), and also the level of IL-6 and TNF-α in culture supernatants. Inhibitors for p38 MAPK, phosphatidylinositol 3-kinase (PI3K) /AKT and NF-κB signaling pathways did not affect the inhibition of Cantharidin on VSMCs proliferation, migration and inflammation. These findings indicated that Cantharidin could significantly inhibit the proliferation, migration and inflammatory response of VSMCs, which suggested that Cantharidin may be a potential inhibitor for neointimal hyperplasia and restenosis after angioplasty.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2550K) HTML形式で全画面表示 -
Toru Kimura, Ai Tsukada, Toshiyuki Fukutomi, Kimiyoshi Ichida, Sumio O ...2019 年42 巻1 号 p. 43-49
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Urate is the final oxidation product of purine metabolism in humans. We have recently reported that the paracellular route is the major urate transport pathway across the blood–placental barrier. In this study, the mechanism of urate paracellular transport was investigated in several epithelial cell lines including Madin–Darby canine kidney (MDCK) type I, Lilly Laboratories cell-porcine kidney 1 (LLC-PK1) and Caco-2 cells. Very little urate passed through MDCK and LLC-PK1 cell layers. In contrast, one of the Caco-2 cell lines was found to be urate-permeable. This urate paracellular movement across Caco-2 cell layer was not inhibited by the urate transporter inhibitor benzbromarone but was partially inhibited by 4,4′-diisothiocyanato-2,2′-stilbenedisulfonic acid (DIDS), which inhibits chloride transport. Detection and quantification of claudin proteins that are important for paracellular transport of ions were performed by LC/MS. Claudins 1, 3, 4, 6, 7 and 12 were detected in urate-permeable cell lines, BeWo cells and Caco-2 cells. We compared claudin expression patterns in urate-permeable and urate-non-permeable Caco-2 cells by LC/MS and found that claudin 12 had a higher expression level in urate-permeable Caco-2 cells. Overexpression of these claudins in MDCK cells did not increase urate paracellular transport. Although there were differences in claudin expression pattern between urate-permeable and non-permeable cells, increased expression of single claudin alone did not explain paracellular permeability of urate.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (928K) HTML形式で全画面表示 -
Cong Ma, Bo Wen, Qin Zhang, Peipei Shao, Wen Gu, Kun Qu, Yang Shi, Bei ...2019 年42 巻1 号 p. 50-56
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/10/17 ジャーナル フリー HTML
ジャーナル フリー HTMLThe development of ankylosing spondylitis (AS) occurs due to excessive proliferation of fibroblasts. Polydatin, a monomeric compound isolated from a traditional Chinese medicine Polygonum cuspidatum, exhibits anti-inflammatory and anti-arthritic effects. However, the mechanisms underlying the regulatory effects of polydatin on the proliferation, apoptosis and autophagy of fibroblasts obtained from patients with AS remain unclear. The aim of this study was to investigate the therapeutic effects of polydatin on symptoms associated with AS. Multiple cellular and molecular biology experiments were performed in the present study, such as cell viability assay, Western blotting, flow cytometry, monodansylcadaverine (MDC) staining and immunofluorescence assays. In the present study, the results revealed that polydatin induced the apoptosis of fibroblasts isolated from patients with AS by upregulating the expression of active caspase-3 and Bax, and downregulating the expression of Bcl-2. Meanwhile, polydatin was revealed to enhance the autophagy of fibroblasts by increasing the expression levels of LC3II, Beclin 1 and Atg5. The results of MDC and immunofluorescence assays further demonstrated that polydatin significantly induced the formation of autophagosomes in fibroblasts. Furthermore, polydatin-induced apoptosis and autophagy were markedly inhibited following treatment with the autophagy inhibitor, 3-methyladenine (3-MA). In conclusion, the results of the present study indicated that polydatin induces the apoptosis and autophagy of fibroblasts obtained from patients suffering from AS, and that polydatin may represent a therapeutic agent for the future treatment of patients with AS.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3964K) HTML形式で全画面表示 -
 Norihito Kikuchi, Jiabin Ye, Jotaro Hirakawa, Hiroto Kawashima2019 年42 巻1 号 p. 57-65
Norihito Kikuchi, Jiabin Ye, Jotaro Hirakawa, Hiroto Kawashima2019 年42 巻1 号 p. 57-65
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/11/01 ジャーナル フリー HTML
ジャーナル フリー HTMLCXC chemokine ligand 10 (CXCL10) is a CXC chemokine family protein that transmits signals by binding to its specific receptor, CXCR3. CXCL10 is also known as an interferon-γ-inducible chemokine involved in various biological phenomena, including chemotaxis of natural killer (NK) cells and cytotoxic T lymphocytes, that suppress tumor growth and inhibition of angiogenesis. In this study, we examined the effects of forced expression of CXCL10 in a murine colon carcinoma cell line (CT26) on growth and metastasis in syngeneic mice. We first established CT26 cells that were stably expressing murine CXCL10 (CT26/CXCL10) and compared their growth with their parental CT26 cells in vitro and in vivo. The in vitro growth of the CT26/CXCL10 and CT26 cells was comparable, whereas the in vivo growth of the CT26/CXCL10 cells in the skin was strongly suppressed. Liver metastasis of the CT26/CXCL10 cells was also significantly suppressed after intra-splenic implantation. Removal of NK cells by the administration of anti-asialo GM1 antibody canceled the suppression of subcutaneous growth and liver metastasis of CT26/CXCL10 cells. Immunofluorescence clearly showed that abundant NKp46-positive NK cells were recruited into the liver metastatic lesions of the CT26/CXCL10 cells, consistent with specific NK cell migration towards the culture supernatant from the CT26/CXCL10 cells in the chemotaxis assay using transwells. These findings indicate that CXCL10 prevents in vivo growth and metastasis of colon carcinoma cells by recruiting NK cells, suggesting that forced expression of CXCL10 in the colon tumors by gene delivery should lead to a favorable clinical outcome.
Graphical Abstract Fullsize Image抄録全体を表示Editor's pickCXC chemokine ligand (CXCL) 10 is a chemokine that binds to CXCR3 expressed on natural killer (NK) cells and cytotoxic T lymphocytes. In this paper, Kikuchi et al. showed that forced expression of CXCL10 in murine colon carcinoma CT26 cells prevents their in vivo proliferation and liver metastasis by recruiting NK cells, suggesting that forced expression of CXCL10 in the colon tumors by gene delivery should lead to a favorable clinical outcome.
PDF形式でダウンロード (10716K) HTML形式で全画面表示 -
Hoyoung Ryu, Hyunjin Jin, Jin-Nyoung Ho, Jungbum Bae, Eunsik Lee, Sang ...2019 年42 巻1 号 p. 66-72
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
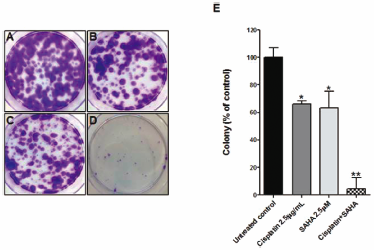
ジャーナル フリー HTMLCisplatin chemotherapy is the standard treatment for metastatic urothelial carcinoma. Although there are second-line chemotherapeutic agents approved by the U.S. Food and Drug Administration (FDA) such as those targeting programmed death-ligand 1 (PD-L1), more effective pharmacotherapy is required for cisplatin-resistant bladder cancer due to its limited overall survival and progression-free survival. The synergistic anti-cancer effect of cisplatin and suberoylanilide hydroxamic acid (SAHA) in cisplatin-resistant bladder cancer cells (T24R2) was examined. Tumor cell proliferation and cell cycle was examined using the cell counting kit (CCK)-8 assays and flow cytometry, respectively. Synergism was examined using the combination index (CI). CCK-8 assay and CI test were used to observe the strong synergistic anti-cancer effect between SAHA and cisplatin. Activation of caspase mediated apoptosis, down-regulated expression of the anti-apoptotic B-cell lymphoma-2 (Bcl-2) and up-regulated expression of pro-apoptotic Bcl-2-associated death promoter (BAD) were observed in Western blot. SAHA synergistically could partially re-sensitize cisplatin-resistant bladder cancer cells (T24R2) through the cell cycle arrest and induction of apoptosis pathway. SAHA-based treatment could be a potential treatment regimen in patients with cisplatin resistant bladder cancer.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1435K) HTML形式で全画面表示 -
Norito Nishiyama, Taro Yamaguchi, Masanori Yoneyama, Yusuke Onaka, Kiy ...2019 年42 巻1 号 p. 73-80
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLIt is well-known that outer hair cell (OHC) loss occurs in the cochlea of animal models of permanent hearing loss induced by intense noise exposure. Our earlier studies demonstrated the production of hydroxynonenal and peroxynitrite, as well as the disruption of gap junction-mediated intercellular communication (GJIC), in the cochlear spiral ligament prior to noise-induced sudden hearing loss. The goal of the present study was to evaluate the mechanism underlying cochlear OHC loss after sudden hearing loss induced by intense noise exposure. In organ of Corti explant cultures from mice, no significant OHC loss was observed after in vitro exposure to 4-hydroxynonenal (a product of lipid peroxidation), H2O2, SIN-1 (peroxynitrite generator), and carbenoxolone (a gap junction inhibitor). Interestingly, in vivo intracochlear carbenoxolone injection through the posterior semicircular canal caused marked OHC and hearing loss, as well as the disruption of gap junction-mediated intercellular communication in the cochlear spiral ligament. However, no significant OHC loss was observed in vivo in animals treated with 4-hydroxynonenal and SIN-1. Taken together, our data suggest that disruption of GJIC in the cochlear lateral wall structures is an important cause of cochlear OHC loss in models of hearing loss, including those induced by noise.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2177K) HTML形式で全画面表示 -
Yuki Yamamoto, Satohiro Masuda, Hiroshi Nakase, Minoru Matsuura, Shiho ...2019 年42 巻1 号 p. 81-86
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/10/24 ジャーナル フリー HTML
ジャーナル フリー HTMLThe efficacy of 5-aminosalicylic acid (5-ASA) as the first-line therapy for ulcerative colitis (UC) is determined by the extent of drug delivery to the inflamed region. Moreover, differences among the various formulations influence delivery of the drug. In this study, we examined the clinical significance of colonic mucosal concentrations of 5-ASA and N-acetylmesalamine (Ac-5-ASA) in UC patients receiving a pH-dependent or time-dependent release formulation of 5-ASA. The subjects were 67 patients with UC who were treated with a pH-dependent or time-dependent formulation of 5-ASA between December 2011 and April 2014. A retrospective observational analysis of clinical outcomes was performed using the clinical activity index (CAI) obtained on the day of biopsy. Colonic mucosal concentrations of 5-ASA and Ac-5-ASA in biopsy samples were measured by LC-tandem mass spectrometry/mass spectrometry. Patients who were treated with the pH-dependent formulation had higher colon mucosal concentrations of 5-ASA than those who were treated with the time-dependent formulation. Additionally, 5-ASA concentration was significantly higher in patients with CAI scores ≤3. A higher concentration of Ac-5-ASA was achieved with the time-dependent formulation than with the pH-dependent formulation. Furthermore, patients with CAI scores ≤3 had higher concentrations of 5-ASA than those with CAI scores ≥4. The colonic mucosal concentration of 5-ASA in patients with UC is influenced by the pharmaceutical formulation and the remission status of UC.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (445K) HTML形式で全画面表示 -
Ryota Uchida, Huihui Xiang, Hiroya Arai, Hidemitsu Kitamura, Keigo Nis ...2019 年42 巻1 号 p. 87-93
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/11/10 ジャーナル フリー HTML
ジャーナル フリー HTMLThe trace element zinc is essential for the immune system, and its dysregulation and deficiency results in impaired immune function. Recent studies have shown that zinc can behave as an intracellular signaling molecule in immune cells. We have previously demonstrated that L-type calcium channel (LTCC) is involved in the regulation of zinc signaling, Zinc wave and cytokine production by stimulating Fc epsilon receptor for immunoglobulin E (IgE) in mast cells. However, it is not known whether LTCC-mediated Zinc wave is required for cytokine production by stimulation of toll-like receptors and cytokine receptors in mast cells. Here we report that stimulation of toll-like receptors and cytokine receptors can induce Zinc wave in mast cells and regulate the expression of cytokine genes. The LTCC antagonist nicardipine inhibited lipopolysaccharide (LPS)- and interleukin-33 (IL-33)-mediated Zinc wave and the induction of cytokine genes such as IL-6. Consistent with these results, the zinc chelator N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) also inhibited LPS- and IL-33-induced cytokine gene expression. Furthermore, LPS induced Zinc wave not only in mast cells but also in dendritic cells. Together, these observations show that Zinc wave is activated by various stimuli and is linked to cytokine gene induction in immune cells.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (766K) HTML形式で全画面表示 -
Euteum Park, Jungsoo Gim, Do Kyung Kim, Chun-Sung Kim, Hong Sung Chun2019 年42 巻1 号 p. 94-102
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLGlutamate-mediated cytotoxicity has been implicated in the pathogenesis of neurological diseases, including Parkinson’s disease, Alzheimer’s disease, and stroke. In this study, we investigated the protective effects of alpha-lipoic acid (ALA), a naturally occurring thiol antioxidant, on glutamate-induced cytotoxicity in cultured C6 astroglial cells. Exposure to high-dose glutamate (10 mM) caused oxidative stress and mitochondrial dysfunction through the elevation of reactive oxygen species, depletion of glutathione, and loss of the mitochondrial membrane potential (ΔΨm). Pretreatment with ALA (200 µM), however, significantly inhibited the glutamate-induced oxidative stress and mitochondrial dysfunction. ALA pretreatment dose-dependently suppressed glutamate-induced apoptotic events including altered nuclear morphology and activation of caspase-3. In addition, ALA significantly attenuated glutamate-induced endoplasmic reticulum (ER) stress markers; namely, glucose-regulated protein 78 (GRP78), activating transcription factor 6 (ATF6), protein kinase regulated by RNA (PKR)-like ER-associated kinase (PERK), eukaryotic translation initiation factor 2 alpha (eIF2α), inositol-requiring enzyme 1 (IRE1), CCAAT/enhancer binding protein homologous protein (CHOP), and caspase-12. We confirmed that CHOP and caspase-12 are key mediators of glutamate-induced ER stress. Furthermore, exposure of the cells to a caspase-12-specific inhibitor and CHOP small interfering RNAs (siRNAs) led to restoration of the ΔΨm that was damaged by glutamate treatment. These results suggest that ALA can effectively suppress oxidative stress, mitochondrial dysfunction, and ER stress in astroglial cells.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3568K) HTML形式で全画面表示 -
Yuji Saito, Tomoaki Usami, Miki Katoh, Masayuki Nadai2019 年42 巻1 号 p. 103-109
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThylakoid-rich spinach extract is being used as dietary weight-loss supplements in Japan. A recent rat study has suggested that intake of thylakoid-rich spinach extract with dietary oil inhibits dietary fat absorption via binding to bile acids, which promotes excretion of bile acids in feces. While, we confirmed that a serving size of thylakoid-rich spinach extract contains a large amount of calcium (130 mg/5 g). Therefore, using rats, we evaluated whether one-time ingestion of thylakoid-rich spinach extract affects the gastrointestinal absorption of water-insoluble drugs, such as griseofulvin (GF) and indomethacin (IM), or ciprofloxacin (CPFX) that chelate with polyvalent metal cations. Pretreatment of the rats with thylakoid-rich spinach extract (100 or 300 mg/kg) for 15 min prior to oral administration of GF (50 mg/kg) or IM (10 mg/kg) did not significantly alter the pharmacokinetic properties of either drug. Meanwhile, co-administration of thylakoid-rich spinach extract (500 mg/kg) and CPFX (20 mg/kg) significantly reduced the peak plasma concentration and the area under the plasma concentration-time curve of CPFX to 25 and 40%, respectively in rats. In vitro studies demonstrated that when a mixture of thylakoid-rich spinach extract and CPFX was centrifuged, there was a significant reduction in the supernatant concentration of CPFX relative to the control. When the experiment was repeated in the presence of ethylenediaminetetraacetic acid, the concentration of CPFX was unchanged. These results suggest that the intake of thylakoid-rich spinach extract may reduce the absorption of drugs that form a chelate with polyvalent metal cations, such as CPFX.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (526K) HTML形式で全画面表示 -
Nanami Shigetomi, Kenta Kamiya, Toru Takamori, Naoki Yoshimura, Sayaka ...2019 年42 巻1 号 p. 110-115
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe purpose of this study was to determine the serum protein binding of tadalafil in children with protein-losing enteropathy (PLE) and to evaluate the specific binding of the drug to human serum-derived proteins in vitro. Seventeen serum samples from two PLE patients used after biochemical tests were collected, and the unbound fraction of tadalafil was determined by an ultrafiltration method. The serum albumin concentrations observed in patients #1 and #2 were 2.4–4.2 and 2.9–3.5 g/dL, respectively. The ranges of unbound fraction of tadalafil in patients #1 and #2 were 3.9–13 and 5.0–7.0%, respectively. This suggested that serum albumin was at least a binding carrier for tadalafil because the unbound fraction of tadalafil and serum albumin were slightly correlated. The unbound fraction of tadalafil at the total concentration of 300 ng/mL was negatively dependent on the serum albumin concentration (range: 1.0–5.0 g/dL) in vitro. In the presence of albumin, the additive effect of γ-globulin on the unbound fraction of tadalafil was marginal, but the addition of α1-acid glycoprotein to test samples decreased the unbound fraction of the drug. The decrease in the unbound fraction of tadalafil was greater at low albumin levels (2 g/dL). The addition of lipoprotein to test samples also decreased the unbound fraction of tadalafil, suggesting that lipoprotein was also a binding carrier of the drug. These results suggested that the disposition and/or response to tadalafil in PLE patients was altered by the change in protein bindings of the drug.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (611K) HTML形式で全画面表示 -
Hiroyuki Taguchi, Haruki Tanaka, Kaname Hashizaki, Yoshihiro Saito, Ma ...2019 年42 巻1 号 p. 116-122
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/10/23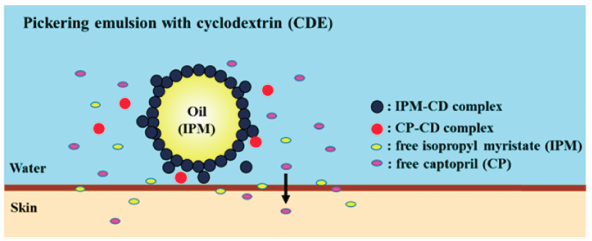 ジャーナル フリー HTML
ジャーナル フリー HTMLThe emulsion prepared with β-cyclodextrin as an emulsifier (βCDE) is considered to be a Pickering emulsion. We examined the characteristics of βCDEs using captopril (CP) as a model drug, and studied the in vitro skin permeation of CP from βCDEs through hairless mouse skin. The stability of βCDE was increased with increasing βCD concentration and conversely decreased with increasing CP concentration. The yield stress value from the rheological measurement results was suggested to be one of the factors determining the stability of the βCDE, and βCDEs with higher yield stress values were more stable. We found that the skin permeability of CP could be improved by using βCDE with isopropyl myristate as the oil phase and that the flux of CP depended on the free CP concentration in the water phase of βCDE.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1451K) HTML形式で全画面表示 -
Guopeng Miao, Juan Han, Jifeng Zhang, Yihai Wu, Guanhe Tong2019 年42 巻1 号 p. 123-129
発行日: 2019/01/01
公開日: 2019/01/01
[早期公開] 公開日: 2018/10/30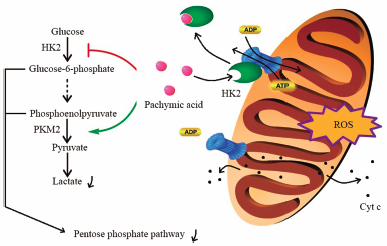 ジャーナル フリー HTML
ジャーナル フリー HTMLPachymic acid (PA), a triterpenoid from Poria cocos, has various pharmacological effects, including anti-inflammatory, anti-cancer, anti-aging, and insulin-like properties. PA has gained considerable research attention, but the mechanism of its anti-cancer effects remains unclear. In this study, pyruvate kinase M2 (PKM2) was discovered as a PA target via the drug affinity responsive target stability. Molecular docking and enzyme assay revealed that PA is a competing activator of PKM2, and mimics the natural activator, fructose-1,6-bisphosphate. PKM2 activation should augment the flux of glycolysis. However, decreased glucose uptake and lactate production after PA treatment was observed in SK-BR-3 breast carcinoma cells, indicating a blockage or downregulation of glycolysis. The potential of previously reported triterpenoids in blocking hexokinase II (HK2) activity inspired us to investigate the inhibition effect of PA on HK2 activity. Molecular docking and enzyme assay confirmed that PA was an inhibitor of HK2, with an IC50 of 5.01 µM. The possible consequences of glycometabolic regulation by PA, such as dissociation of HK2 from the mitochondria, release of mitochondrial cytochrome (Cyt) c, depletion of ATP, and generation of reactive oxygen species, were further validated. Furthermore, the details of the possible linkage of targeting PKM2 and HK2 with previously reported actions of PA were discussed. The results of our study provided valuable information on the anti-cancer mechanisms of PA.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2132K) HTML形式で全画面表示 -
Koichi Hamada, Yosuke Maeda, Akihiro Mizutani, Seiji Okada2019 年42 巻1 号 p. 130-138
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
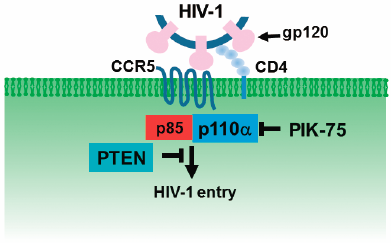
ジャーナル フリー HTMLHuman immunodeficiency virus type 1 (HIV-1) drives multiple signaling pathways to facilitate its cellular entry and replication. The interaction between HIV-1 envelope (env) protein and target cell surface CD4 first activates the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, and the subsequent interaction between HIV-1 env glycoprotein and CCR5/CXCR4 coreceptors establishes viral fusion and entry. Four isoforms of the class-I PI3K catalytic subunits (p110α, p110β, p110γ, and p110δ) have been identified so far, but the isoform(s) involved in the HIV-1 entry is still unknown. This study aimed to identify the PI3K isoform(s) using recently developed isoform-specific inhibitors and the roles of their negative regulators, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1), in HIV-1 infection. We found that the PI3K p110α isoform-specific inhibitor PIK-75 suppressed HIV-1 entry in HIV-1 permissive T cells, PM1 cells, and TZM-bl cells (HeLa cell-derived indicator cells that coexpress CD4, CCR5, and CXCR4) and decreased the HIV-1-induced phosphorylation of Akt. Moreover, wild-type PTEN (but neither phosphatase-deficient PTEN nor wild-type SHIP1) was a key regulator of HIV-1 entry. Cell-to-cell fusion by HIV-1 env–CD4 interaction was suppressed in the presence of PI3K p110α-specific inhibitor. These data suggest that the PI3K p110α/PTEN signaling pathway is indispensable for HIV-1 entry, including HIV-1 env-mediated cell-to-cell fusion.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2407K) HTML形式で全画面表示
-
Hoon-Seok Yoon, Jung-Il Kang, Sung Min Kim, Ara Ko, Young-Sang Koh, Ji ...2019 年42 巻1 号 p. 139-143
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Norgalanthamine has been shown to possess hair-growth promoting effects, including increase in hair-fiber length in cultured rat vibrissa follicles and increase in dermal papilla cell (DPC) proliferation. However, the intracellular mechanisms that underlie the action of norgalanthamine in DPCs have not been investigated. In this study, we addressed the ability of norgalanthamine to trigger anagen-activating signaling pathways in DPCs. Norgalanthamine significantly increased extracellular signal-regulated kinase (ERK) 1/2 phosphorylation at 0.1 µM, a concentration at which DPC proliferation was also induced. Furthermore, the increases in norgalanthamine-induced ERK 1/2 activation and subsequent DPC proliferation were suppressed by the mitogen-activated protein kinase/ERK kinase (MEK) 1/2 inhibitor, U0126. A 0.1 µM dose of norgalanthamine also increased phosphorylation of AKT, which was followed by an increase in glycogen synthase kinase 3β phosphorylation and nuclear translocation of β-catenin. In addition, LY294002, a phosphatidylinositol 3 kinase (PI3K) inhibitor, blocked the effect of norgalanthamine on DPC proliferation. These results suggest that norgalanthamine can stimulate the anagen phase of the hair cycle in DPCs via activation of the ERK 1/2, PI3K/AKT, and Wnt/β-catenin pathways.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (682K) HTML形式で全画面表示 -
Shigehiro Omori, Yusuke Kamiya, Tsutomu Yamaki, Masaki Uchida, Kazuo O ...2019 年42 巻1 号 p. 144-148
発行日: 2019/01/01
公開日: 2019/01/01
 ジャーナル フリー HTML
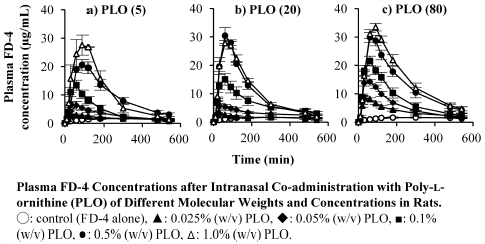
ジャーナル フリー HTMLThe transnasal route for the delivery of water-soluble macromolecules, such as bioactive peptides and proteins, has attracted interest, although the use of permeation enhancers is required due to the poor permeabilities of these macromolecules across the nasal mucosa. With polycationic compounds, such as poly-L-arginine and chitosan, the nasal absorption of hydrophilic macromolecules is molecular weight- and concentration-dependently enhanced without causing cytotoxicity. In the present study, we evaluated the effect of various molecular weights and concentrations of poly-L-ornithine (PLO), a polycationic compound, on the nasal absorption and the damage to the nasal mucosa in vivo. PLO enhanced the nasal absorption of fluorescein isothiocyanate-dextran (FD-4), used as a model drug, and the bioavailability of FD-4 increased with the concentration of PLO. The enhancement effect was also dependent on the molecular weight. The administration of PLO at a concentration that sufficed for enhancing the nasal absorption had no effect on the activity of lactic dehydrogenase and the protein leakage in the nasal fluid, as indices of nasal mucosa damage. These findings suggest that a transnasal delivery system using PLO is a useful strategy for improving the nasal absorption of water-soluble macromolecules without toxicity to the nasal mucosa.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (455K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|