
- |<
- <
- 1
- >
- >|
-
Naoto Oku2017 年 40 巻 2 号 p. 119-127
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLLiposomes have been widely used as drug carriers in the field of drug delivery systems (DDS), and they are thought to be ideal nano-capsules for targeting DDS after being injected into the bloodstream. In general, DDS drugs meet the needs of aged and super-aged societies, since the administration route of drugs can be changed, the medication frequency reduced, the adverse effects of drugs suppressed, and so on. In fact, a number of liposomal drugs have been launched and used worldwide including liposomal anticancer drugs, and these drugs have appeared on the market owing to various innovations in liposomal DDS technologies. The accumulation of long-circulating liposomes in cancer tissue is driven by the enhanced permeability and retention (EPR) effect. In this review, liposome-based targeting DDS for cancer therapy is briefly discussed. Since cancer angiogenic vessels are the ideal target of drug carriers after their injection and are critical for cancer growth, damaging of these neovessels has been an approach for eradicating cancer cells. Also, the usage of liposomal DDS for the treatment of ischemic stroke is possible, since we observed that PEGylated liposomes accumulate in the site of cerebral ischemia in transient middle cerebral artery occlusion (t-MCAO) model rats. Interestingly, liposomes carrying neuroprotectants partly suppress ischemia/reperfusion injury of these model rats, suggesting that the EPR effect also works in ischemic diseases by causing an increase in the permeability of the blood vessel endothelium. The potential of liposomal DDS against life-threatening diseases might thus be attractive for supporting long-lived societies.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (5155K) HTML形式で全画面表示 -
Yu Ishima2017 年 40 巻 2 号 p. 128-134
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLBiomacromolecules (>40 kDa) have been developed as drug delivery system (DDS) carriers of low-molecular weight drugs to promote these drugs’ uptake by cancer tissues via enhanced permeability and retention (EPR) effects. Human serum albumin (HSA) has been found to accumulate in cancer tissues via this EPR effect. HSA is the most abundant protein in serum, which performs essential physiological functions such as the transportation of many endogenous and exogenous ligands. Nitric oxide (NO) is a very small ligand of HSA; it is a unique and diffusible molecular messenger that plays a central role in mammalian physiology. Although the in vivo half-life of NO is extremely short, HSA could prolong the half-life of NO via S-nitrosation at the position of Cys-34. S-Nitrosated HSA (mono-SNO-HSA) is called an ‘Endogenous NO traffic protein,’ due to the highly stable S-nitroso form in circulating blood, and to the efficiency of S-transnitrosation in cells that require NO. Mono-SNO-HSA possesses a very strong cytoprotective action via the induction of heme oxygenase-1. On the other hand, HSA reinforced with approximately seven NO molecules (poly-SNO-HSA), which we developed by means of chemical modification, possesses multiple anticancer activities. Our previous data clarified that the high expression of protein disulfide isomerase on the surface of cancer cells plays a very important role in the anticancer action of poly-SNO-HSA. In this review, we focus on the advantage of poly-SNO-HSA in treating intractable cancers from the viewpoint of drug delivery systems and drug resistance.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (4452K) HTML形式で全画面表示
-
Yadan Zhang, Lijuan Song, Ruiyan Pan, Jianwei Gao, Bao-xia Zang, Ming ...2017 年 40 巻 2 号 p. 135-144
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLHydroxysafflor yellow A (HSYA) is an effective ingredient of the Chinese herb Carthamus tinctorius L. The present study investigated the protective effect of HSYA on lipopolysaccharide (LPS)-induced acute respiratory distress syndrome in mice, and the underlying mechanisms involved. HSYA (14, 28, 56 mg/kg) was intraperitoneally injected to mice once daily from day 1 to 10 after LPS administration. HSYA attenuated the body weight loss, the augmented left index and the increase of pathologic changes in pulmonary inflammation caused by LPS. Treatment with HSYA also alleviated increased expressions of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, transforming growth factor (TGF)-β1, collagen (Col) I, Col III, α-smooth muscle actin (α-SMA), myeloid differentiation (MD)-2, Toll-like receptor 4 (TLR4) and cluster differentiation (CD)14 at the mRNA (RT-PCR) and protein levels (Western blot and enzyme-linked immuno sorbent assay). Moreover, HSYA inhibited the elevated levels of nuclear factor (NF)-κB and α-SMA in lung tissue (immunohistochemistry), and alleviated the slight collagen deposition in pulmonary tissues (Masson’s trichrome staining). HSYA inhibited the specific binding of fluorescein isothiocyanate (FITC)-LPS on human lung epithelial cell line (A549) or human umbilical vein cell line (Eahy926) cells (flow cytometry). These findings suggested that HSYA has a protective effect on acute respiratory distress syndrome (ARDS) induced by LPS through blocking the TLR4/NF-κB pathway, and that the TLR4 receptor might be a target of HSYA on the cell membrane.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (5058K) HTML形式で全画面表示 -
Yusuke Tanetsugu, Tatsuaki Tagami, Takayuki Terukina, Takaya Ogawa, Ma ...2017 年 40 巻 2 号 p. 145-150
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLRanibizumab is a humanized monoclonal antibody fragment against vascular endothelial growth factor (VEGF)-A and is widely used to treat age-related macular degeneration (AMD) caused by angiogenesis. Ranibizumab has a short half-life in the eye due to its low molecular weight and susceptibility to proteolysis. Monthly intravitreal injection of a large amount of ranibizumab formulation is a burden for both patients and medical staff. We therefore sought to develop a sustainable release system for treating the eye with ranibizumab using a drug carrier. A ranibizumab biosimilar (RB) was incorporated into microparticles of poly(lactic-co-glycolic acid) (PLGA) biodegradable polymer. Ranibizumab was sustainably released from PLGA microparticles (80+% after 3 weeks). Assay of tube formation by endothelial cells indicated that RB released from PLGA microparticles inhibited VEGF-induced tube formation and this tendency was confirmed by a cell proliferation assay. These results indicate that RB-loaded PLGA microparticles are useful for sustainable RB release and suggest the utility of intraocular sustainable release systems for delivering RB site-specifically to AMD patients.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1972K) HTML形式で全画面表示 -
Yoon Ah Sohn, In Young Hwang, Sun Yi Lee, Hyo Sun Cho, Choon Sik Jeong2017 年 40 巻 2 号 p. 151-154
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
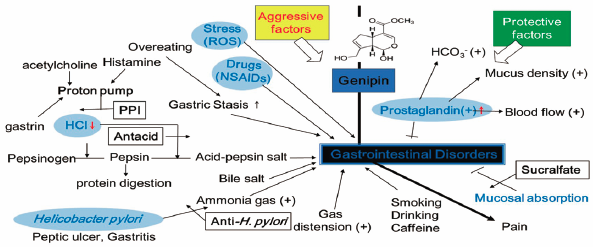
ジャーナル フリー HTMLGenipin, an aglycone of geniposide, is a major component of gardeniae fructus, and has been used to treat jaundice, various inflammatory disorders, and liver disease, and has also been used as a natural cross-linking agent. The authors conducted several experiments to evaluate the protective effects of genipin on gastrointestinal disorders, such as, gastritis and gastric ulcers. Genipin showed inhibitory effects against HCl·ethanol-induced acute gastritis and indomethacin-induced gastric ulcers in rats and increased prostaglandin E2 (PGE2) in AGS gastric cancer cell. Genipin had significant effects on aggressive factors, acid-neutralization, and gastric secretion, and inhibited H+/K+-ATPase (a proton pump), which secretes gastric acid. The results obtained indicate that genipin has significant gastroprotective effects and might be useful for treating and preventing gastric lesions.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (303K) HTML形式で全画面表示 -
Shihomi Hirooka, Misaki Ueno, Satoko Fukuda, Atsushi Miyajima, Takashi ...2017 年 40 巻 2 号 p. 155-160
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLIn the present study, the relationship between systemic exposure of simvastatin (SV) hydroxy acid (SV-acid), an active form of SV, and its alveolar regeneration rates was investigated using emphysema model mice created by postnatal treatment of dexamethasone. In a model with young animals, the mice were treated with SV for 10 d from postnatal day 42. Similar alveolar regeneration with a % mean linear intercept (Lm) recovery of 60 to 70% by histochemical observation was observed in mice after intraperitoneal administration at dose in the range of 4–100 µg/mouse. The % Lm recovery after oral administration of 20 µg/mouse was comparable with that after intraperitoneal administration at a dose of 4 µg/mouse, when their exposure of SV-acid was almost similar in both treated groups. Regardless of the route of administration, the recovery can depend on the exposure level of SV-acid, and to the maximum was about 60–70%. On the other hand, in a model with adult animals, the mice were intraperitoneally administrated SV at a dose of 4 µg/mouse for 10 d from postnatal day 152. Compared to young animals, less % Lm recovery was observed in adult mice even their systemic exposures of SV-acid were similar.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (912K) HTML形式で全画面表示 -
Naho Maruyama, Shigeru Tansho-Nagakawa, Chizuru Miyazaki, Kazuyuki Shi ...2017 年 40 巻 2 号 p. 161-168
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
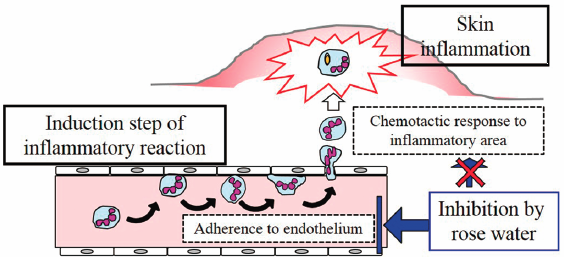
電子付録Hydrosol prepared from the flowers of Rosa damascena (rose water) has been traditionally used for various health-related issues, including skin troubles such as erythema, itchiness, swelling. For the care of these skin troubles caused by microbial infection, both antimicrobial and antiinflammatory effects are required. Here, we investigated the effects of rose water on the growth of Candida albicans and methicillin-resistant Staphylococcus aureus (MRSA), which cause skin infections, and on the function of neutrophils, which play a major role in the regulation of inflammatory reactions. To assess its modulatory effects on neutrophils, the effects of rose water against neutrophil adhesion response were evaluated. Rose water inhibited mycelial growth of C. albicans at a concentration of ca. 2.2%, and reduced viability of MRSA within 1 h. Rose water suppressed neutrophil activation induced by lipopolysaccharide (LPS), tumor necrosis factor alpha (TNF-α), and N-formyl-Met-Leu-Phe (fMLP) at 5–15%. It also reduced the LPS- and TNF-α-induced cell surface expression of the adhesion-related molecule, cluster of differentiation (CD) 11b, but did not affect the migratory capacity of neutrophils with or without chemoattractant. These results suggest that rose water may reduce the pathogenicity of microbes, and attenuate neutrophil stimulation, which is involved in inflammatory responses. These findings suggest that rose water has a potential effect to inhibit skin inflammation caused by microbes.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1662K) HTML形式で全画面表示 -
Liang He, Jun-Mei Xu, Su-Mei Liu, Zhi-Jun Chen, Xin Li, Rong Zhu2017 年 40 巻 2 号 p. 169-173
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLShivering associated with spinal anesthesia during Cesarean delivery is an uncomfortable experience for the parturient, which may also cause adverse effects. In this prospective, randomized, double-blind, placebo-controlled study, we sought to evaluate the effect of intrathecal dexmedetomidine, administered as an adjunct to hyperbaric bupivacaine for Cesarean delivery, on the incidence and severity of shivering associated with spinal anesthesia. Patients undergoing Cesarean delivery were randomly allocated to three groups of 30 patients each. Experimental treatments were added to hyperbaric bupivacaine as follows: Patients in group I (control) were administered isotonic saline. Patients in groups II and III received dexmedetomidine (2.5, 5 µg, respectively), mixed with isotonic saline. Shivering was observed in 11, 10 and 2 patients in groups I, II and III, respectively. The incidence of shivering in group III was significantly lower than that in groups I (p=0.005) and II (p=0.01). The severity of shivering was significantly different between the three groups (p=0.01). There were no significant inter-group differences with respect to mean arterial pressure and heart rate at any time point after administration of intrathecal local anesthesia (p>0.05). Intrathecal dexmedetomidine (5 µg) administered as an adjunct to hyperbaric bupivacaine during Cesarean delivery significantly reduced the incidence and intensity of shivering associated with spinal anesthesia.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (519K) HTML形式で全画面表示 -
Izumi Morita, Hiroyuki Oyama, Mayumi Yasuo, Kazuhisa Matsuda, Kengo Ka ...2017 年 40 巻 2 号 p. 174-181
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLLaw enforcement against illicit use of cannabis and related substances requires rapid, feasible, and reliable tools for on-site testing of cannabinoids. Notably, methods based on cannabinoid-specific antibodies enable efficient screening of multiple specimens. Antibody engineering may accelerate development of modern and robust testing systems. Here, we used in vitro affinity maturation to generate a single-chain Fv fragment (scFv) that recognizes with high affinity the psychoactive cannabinoid, Δ9-tetrahydrocannabinol (THC). A mouse monoclonal antibody against THC, Ab-THC#33, with Ka 6.2×107 M−1 (as Fab fragment) was established by the hybridoma technique. Then, a “wild-type” scFv (wt-scFv) with Ka, 1.1×107 M−1 was prepared by bacterial expression of a fusion gene combining the VH and VL genes for Ab-THC#33. Subsequently, random point mutations in VH and VL were generated separately, and the resulting products were assembled into mutant scFv genes, which were then phage-displayed. Repeated panning identified a mutant scFv (scFv#m1-36) with 10-fold enhanced affinity (Ka 1.1×108 M−1) for THC, in which only a single conservative substitution (Ser50Thr) was present at the N-terminus of the VH-complementarity-determining region 2 (CDR2) sequence. In competitive enzyme-linked immunosorbent assay (ELISA), the mutant scFv generated dose–response curves with midpoint 0.27 ng/assay THC, which was 3-fold lower than that of wt-scFv. Even higher reactivity with a major THC metabolite, 11-nor-9-carboxy-Δ9-tetrahydrocannabinol, indicated that the mutant scFv will be useful for testing not only THC in confiscated materials, but also the metabolite in urine. Indeed, the antibody fragment is potentially suitable for use in advanced on-site testing platforms for cannabinoids.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2310K) HTML形式で全画面表示 -
Atsuyuki Saisyo, Rima Shimono, Shigeharu Oie, Kazuhiro Kimura, Hiroyuk ...2017 年 40 巻 2 号 p. 182-186
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLMultiple-dose ophthalmic preparations that do not contain preservatives carry high risks of microbial contamination. However, there are various types of hospital preparations, with different physicochemical properties. In the present study, we evaluated the association between physicochemical properties and microbial contamination in ophthalmic preparations. The investigated hospital preparations included ophthalmic preparations of physiological saline, 0.2% fluconazole, 0.5% vancomycin hydrochloride, and 2% cyclosporine. We investigated the microbial dynamics of each ophthalmic preparation and microbial contamination in ophthalmic preparations used by patients. Remarkable growth of Pseudomonas aeruginosa, Burkholderia cepacia, and Serratia marcescens was observed in ophthalmic preparations of physiological saline and 0.2% fluconazole. All tested microorganisms displayed decreased counts after inoculation in 0.5% vancomycin hydrochloride. In 2% cyclosporine, all investigated microorganisms were below the limit of detection after inoculation for 6 h. The microbial contamination rates of ophthalmic preparations used by patients were 16.7% (3/18 samples) for 0.5% vancomycin hydrochloride and 0% (0/30 samples) for 2% cyclosporine. All detected contaminants in 0.5% vancomycin hydrochloride were Candida spp., one of which was present at a level of 1×104 colony-forming units/mL. The storage method for in-use ophthalmic preparations should be considered on the basis of their physicochemical properties.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (552K) HTML形式で全画面表示 -
Hong-Bo Zhao, You-Ming Jiang, Xiao-Juan Li, Yue-Yun Liu, Xiao-Hui Bai, ...2017 年 40 巻 2 号 p. 187-194
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe current study evaluated the effects of Xiao Yao San (XYS) on anxiety-like behaviors and sought to determine whether the c-Jun N-terminal kinase (JNK) signaling pathway is involved. A total of 40 rats were divided into 5 groups (n=8): the control group (deionized water, per os (p.o.)), the model group (deionized water, p.o.), the SP600125 group (surgery), the per se group (surgery), and the XYS group (3.9 g/kg/d, p.o.). A 1% dimethyl sulfoxide (DMSO) citrate buffer solution (2 µL/ventricle/d) and SP600125 (10 µg/ventricle, 2 µL/ventricle/d) were separately and bilaterally injected into the rats of the two surgery groups via the ventricular system of the brain. All but the control group underwent 14 d of chronic immobilization stress (CIS; 3 h/d). On day 15, the body weights of all of the rats were measured; additionally, the rats were subjected to the elevated plus maze (EPM) and novelty suppressed feeding (NSF) tests. Finally, JNK signaling pathway indices, including phosphorylated JNK (P-JNK), JNK, phosphorylated c-Jun (P-c-Jun) and cytochrome C (Cyt-C), were examined. After modeling, the body weight and behavioral analyses of the model rats indicated that this modeling method induced anxiety-like behaviors. P-JNK, JNK, and P-c-Jun were altered in the hippocampus of the model rats. After 14 d of treatment with XYS and SP600125, rat body weight and behaviors as well as P-JNK, JNK, and P-c-Jun had changed. However, no significant difference in Cyt-C was found. XYS improves the anxiety-like behaviors induced by CIS, which might be related to the JNK signaling pathway in the hippocampus.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (876K) HTML形式で全画面表示 -
Takamasa Hirai, Yoshiaki Yamagishi, Naoya Koizumi, Miwa Nonaka, Rina M ...2017 年 40 巻 2 号 p. 195-204
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLCell-penetrating peptides (CPPs) have been highly anticipated as an efficient delivery system due to their ability to cross biological membranes and transport various cargoes into cells. In the present study, we have identified adenovirus-derived CPPs using various capsid-mutant adenovirus (Ad) vectors. First, we examined the endocytosis-inducing ability of these vectors. A fiber-shaft substituted Ad vector, Ad type 5 vector with the fiber shaft domain replaced by that derived from Ad type 35, induced the highest fluorescein isothiocyanate (FITC)-dextran uptake into a human liver cell line, HepG2 cells. In contrast, the FITC-dextran uptake in HepG2 cells was not significantly different between coxsackievirus and adenovirus receptor (CAR)-binding-ablated Ad vector, integrin-binding-ablated Ad vector or conventional Ad vector. Next, we produced a recombinant Ad type 35 shaft protein using the Escherichia coli recombinant system. The recombinant Ad type 35 shaft protein retained the ability for FITC-dextran uptake and efficient gene delivery by plasmid transfection reagent. Furthermore, we identified 26 C-terminal amino acids in the Ad type 35 shaft protein as the cell membrane binding domain. The 26 amino-acid peptides also have the potential to be internalized into cultured cells. The internalization ability of the peptide was dependent on degree and was inhibited by an actin polymerization inhibitor (Latrunculin B) and by a lipid raft formation inhibitor (methyl-β-cyclodextrin). The results of the present study indicate that Ad type 35-derived peptides induce endocytosis in cultured cells and have the ability to cross biological membranes. This report is the first paper to identify Ad-derived CPPs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2813K) HTML形式で全画面表示 -
Yusuke Kamiya, Tsutomu Yamaki, Masaki Uchida, Tomomi Hatanaka, Mitsuto ...2017 年 40 巻 2 号 p. 205-211
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLPolycationic compounds, such as poly-L-arginine and poly-L-ornithine (PLO), enhance the nasal absorption of hydrophilic macromolecular drugs. However, the bio availability corresponding to the dose of these enhancers has not been obtained in an open system study, where an administered solution is transferred to the pharynx because they do not exhibit mucoadhesion/retention in the nasal cavity. In this study, we prepared PEGylated-poly-L-ornithine (PEG-PLO) and investigated the effects of PEGylation on in vitro adhesion/retention properties, permeation enhancement efficiency, and cytotoxicity. PEG-PLO bearing 3–4 polyethylene glycol (PEG) chains per PLO molecule was more retentive than unmodified PLO on an inclined plate. The permeability of a model drug, FD-4, across Caco-2 cell sheets was enhanced by PEG-PLO as well as by PLO. PLO showed cytotoxicity at high concentrations, whereas PEG-PLO did not decrease cell viability, even above the concentration giving a sufficient enhancement effect. These findings suggest that PEGylation of polycationic absorption enhancers improves their adhesion/retention and decreases their cytotoxicity, which may lead to enhancers with greater utility.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (659K) HTML形式で全画面表示 -
Akiko Tanaka, Tomoyuki Furubayashi, Yuki Enomura, Tomoki Hori, Rina Sh ...2017 年 40 巻 2 号 p. 212-219
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe effect of changes in the mucosal fluid volume on the nasal drug absorption of powder formulations was evaluated using warfarin (WF), piroxicam (PXC), and norfloxacin (NFX) as model drugs. Lactose and sodium chloride (NaCl), which are water soluble and small-sized chemicals that increase osmotic pressure after dissolution, were used as excipients to change the mucosal fluid volume. The in vitro study using a Madin–Darby canine kidney (MDCK) cell monolayer indicated that lactose and NaCl, sprayed over the surface of air interface monolayers, increased the fluid volume on the monolayer surface and enhanced the transepithelial transport of the model drugs. The in vivo animal study indicated that the nasal absorption of PXC is enhanced by lactose and NaCl after nasal administration of the powder formulations. This is likely due to the enhanced dissolution of PXC on fluid-rich nasal mucosa and an increase in the effective surface area for drug permeation, which lead to better nasal absorption. However, both excipients failed to increase the nasal absorption of WF and NFX. To clarify the mechanism of the drug-dependent effect of lactose and NaCl, the nasal residence of the formulation was examined using FD70 as a non-absorbable marker. The nasal clearance of FD70 was enhanced by lactose and NaCl, leading to a decrease in the nasal drug absorption. Lactose and NaCl caused no damage to the nasal tissue. These results indicate that the addition of water-soluble excipients such as lactose to powder formulations can enhance the nasal absorption of highly permeable but poorly soluble drugs.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1015K) HTML形式で全画面表示 -
Hayato Iino, Makiko Fujii, Manami Fujino, Shizuka Kohara, Kaname Hashi ...2017 年 40 巻 2 号 p. 220-226
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
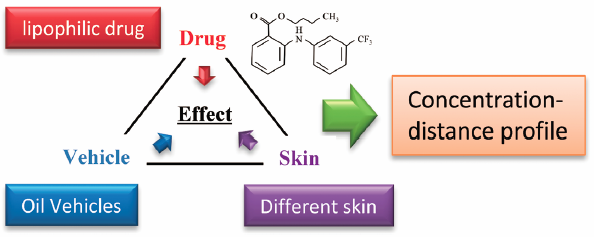
ジャーナル フリー HTMLSkin penetration amounts of a highly lipophilic drug, ufenamate, prepared in four oily vehicles, including white petrolatum (WP), liquid paraffin (LP), isopropyl myristate (IPM), and isocetyl stearate (ICS), were compared. Ufenamate was mixed in each vehicle at 5% and applied at a rate of 2 mg/cm2 to intact, stripped, and delipidized Yucatan micropig skin. The amounts of ufenamate and IPM in the stratum corneum (SC), epidermis, and dermis were determined. The skin penetration amounts of ufenamate from liquid oils were significantly higher than those from WP; the amounts of ufenamate were in the order WP<LP≤ICS<IPM, which was the same as that of the vehicle viscosities. The IPM skin penetration amount was approximately 20 times that of ufenamate. The skin penetration amounts of ufenamate from the liquid vehicles decreased after application to delipidized skin and were not significantly different among the four vehicles. The skin penetration amounts of the vehicle oils were significant and might disrupt intercellular lipid structures, especially in the strips 1–6 of the SC. In the deeper SC, there was no effect of the vehicle or skin condition. Thus, ufenamate mixed in liquid vehicles was found to be an effective dosage form.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (597K) HTML形式で全画面表示
-
Akihiro Fukushima, Wakana Sekiguchi, Kizuku Mamada, Yumi Tohma, Hideki ...2017 年 40 巻 2 号 p. 227-233
発行日: 2017/02/01
公開日: 2017/02/01
[早期公開] 公開日: 2016/12/03 ジャーナル フリー HTML
ジャーナル フリー HTMLAcetaminophen (AcAP), a widely-used antipyretic and analgesic drug, has been considered to exert its effects via central mechanisms, and many studies have demonstrated that the analgesic action of AcAP involves activation of the serotonergic system. Although the serotonergic system also plays an important role in thermoregulation, the contribution of serotonergic activity to the hypothermic effect of AcAP has remained unclear. In the present study, we examined whether the serotonergic system is involved in AcAP-induced hypothermia. In normal mice, AcAP (300 mg/kg, intraperitoneally (i.p.)) induced marked hypothermia (ca. −4°C). The same dose of AcAP reduced pain response behavior in the formalin test. Pretreatment with the serotonin synthesis inhibitor DL-p-chlorophenylalanine (PCPA, 300 mg/kg/d, i.p., 5 consecutive days) substantially decreased serotonin in the brain by 70% and significantly inhibited the analgesic, but not the hypothermic action of AcAP. The same PCPA treatment significantly inhibited the hypothermia induced by the selective serotonin reuptake inhibitor fluoxetine hydrochloride (20 mg/kg, i.p.) and the serotonin 5-HT2 receptor antagonist cyproheptadine hydrochloride (3 mg/kg, i.p.). The lower doses of fluoxetine hydrochloride (3 mg/kg, i.p.) and cyproheptadine hydrochloride (0.3 mg/kg, i.p.) did not affect the AcAP-induced hypothermia. These results suggest that, in comparison with its analgesic effect, the hypothermic effect of AcAP is not mediated by the serotonergic system.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (895K) HTML形式で全画面表示 -
Takashi Nakamura, Yosuke Noma, Yu Sakurai, Hideyoshi Harashima2017 年 40 巻 2 号 p. 234-237
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
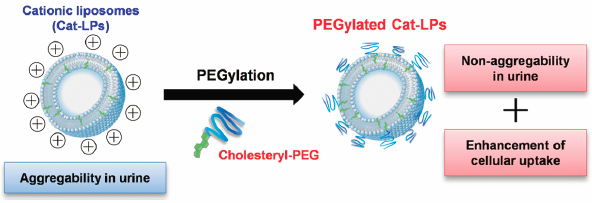
電子付録Intravesical drug delivery by cationic liposomes (Cat-LPs) represents a potent nanotechnology for enhancing therapeutic effects against bladder disorders. However, preventing the aggregation of Cat-LPs in urine poses a significant barrier. We report on an examination of the effect of modifying liposomes with polyethylene glycol (PEG) lipids to prevent Cat-LPs from aggregating in human urine. Although Cat-LPs underwent significant aggregation in human urine, introducing 5 mol% of PEG2k lipid or 2 mol% of PEG5k lipid completely inhibited the aggregation of the Cat-LPs. When 2 mol% of PEG2k lipids were introduced, the lipid structures of 1,2-distearoly-sn-glycero-3-phosphoethanolamine (DSPE) and 1,2-distearoyl-sn-glycerol (DSG) greatly prevented aggregation compared with cholesterol. By contrast, when Cat-LPs, after incubation in urine, were exposed to bladder cancer cells, only introducing cholesteryl-PEG into the Cat-LPs showed a significant enhancement in cellular uptake. These results offer the potential for incorporating cholesteryl-PEG into Cat-LPs for achieving both stability in urine and effective cellular uptake.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (475K) HTML形式で全画面表示 -
Asma Ben Hmidene, Mizuho Hanaki, Kazuma Murakami, Kazuhiro Irie, Hirok ...2017 年 40 巻 2 号 p. 238-241
発行日: 2017/02/01
公開日: 2017/02/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録The prevention of amyloid aggregation is promising for the treatment of age-related diseases such as Alzheimer’s (AD) and type 2 diabetes (T2D). Ten antioxidant flavonoids isolated from the medicinal halophyte Tamarix gallica were tested for their amyloid aggregation inhibition potential. Glucuronosylated flavonoids show relatively strong inhibitory activity of Amyloid β (Aβ) and human islet amyloid polypeptide (hIAPP) aggregation compared to their aglycone analogs. Structure–activity relationship of the flavonoids suggests that the catechol moiety is important for amyloid aggregation inhibition, while the methylation of the carboxyl group in the glucuronide moiety and of the hydroxyl group in the aglycone flavonoids decreased it.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1555K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|