
- |<
- <
- 1
- >
- >|
-
Amr Selim Abu Lila, Tatsuhiro Ishida2017 年 40 巻 1 号 p. 1-10
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe liposome, a closed phospholipid bilayered vesicular system, has received considerable attention as a pharmaceutical carrier of great potential over the past 30 years. The ability of liposomes to encapsulate both hydrophilic and hydrophobic drugs, coupled with their biocompatibility and biodegradability, make liposomes attractive vehicles in the field of drug delivery. In addition, great technical advances such as remote drug loading, triggered release liposomes, ligand-targeted liposomes, liposomes containing combinations of drugs, and so on, have led to the widespread use of liposomes in diverse areas as delivery vehicles for anti-cancer, bio-active molecules, diagnostics, and therapeutic agents. In this review, we summarize design optimization of liposomal systems and invaluable applications of liposomes as effective delivery systems.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1734K) HTML形式で全画面表示
-
Tomoyoshi Miyamoto, Yoshinori Funakami, Erika Kawashita, Ai Nomura, Na ...2017 年 40 巻 1 号 p. 11-16
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
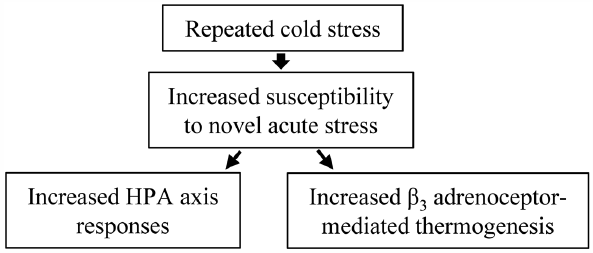
ジャーナル フリー HTMLThe rodents exposed to repeated cold stress according to a specific schedule, known as specific alternation of rhythm in temperature (SART), exhibit autonomic imbalance, and is now used as an experimental model of fibromyalgia. To explore the susceptibility of SART-stressed animals to novel acute stress, we tested whether exposure of mice to SART stress for 1 week alters the extent of acute restraint stress-induced hyperthermia. Mice were subjected to 7-d SART stress sessions; i.e., the mice were alternately exposed to 24 and 4°C at 1-h intervals during the daytime (09:00–16:00) and kept at 4°C overnight (16:00–09:00). SART-stressed and unstressed mice were exposed to acute restraint stress for 20–60 min, during which rectal temperature was monitored. Serum corticosterone levels were measured before and after 60-min exposure to restraint stress. SART stress itself did not alter the body temperature or serum corticosterone levels in mice. Acute restraint stress increased the body temperature and serum corticosterone levels, both responses being greater in SART-stressed mice than unstressed mice. The enhanced hyperthermic responses to acute restraint stress in SART-stressed mice were significantly attenuated by SR59230A, a β3 adrenoceptor antagonist, but unaffected by diazepam, an anxiolytic, mifepristone, a glucocorticoid receptor antagonist, or indomethacin, a cyclooxygenase inhibitor. These results suggest that SART stress enhances the susceptibility of mice to acute restraint stress, characterized by increased hyperthermia and corticosterone secretion, and that the increased hyperthermic responses to acute stress might involve accelerated activation of sympathetic β3 adrenoceptors, known to regulate non-shivering thermogenesis in the brown adipose tissue.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (655K) HTML形式で全画面表示 -
Fang-qiang Zhu, Min-jia Chen, Ming Zhu, Rong-seng Zhao, Wei Qiu, Xiang ...2017 年 40 巻 1 号 p. 17-24
発行日: 2017/01/01
公開日: 2017/01/01
[早期公開] 公開日: 2016/11/08 ジャーナル フリー HTML
ジャーナル フリー HTMLCurcumin has exhibited a protective effect against development of renal fibrosis in animal models, however, its underlying molecular mechanisms are largely unclear. Therefore, we investigated the anti-fibrosis effects of curcumin in transforming growth factor-β1 (TGF-β1)-induced epithelial-to-mesenchymal transition (EMT), and the mechanism by which it mediates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. Human kidney tubular epithelial cells (HKCs) were treated with TGF-β1 or curcumin alone, or TGF-β1 in combination with curcumin. The effect of curcumin on cell proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Expression of E-cadherin, cytokeratin, vimentin, alpha smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1) and key proteins of Akt/mammalian target of rapamycin (mTOR) pathway were analyzed by immunocytochemistry, real-time PCR and Western blot. Low dose curcumin (3.125 and 25 µmol/L) effectively promoted HKC proliferation. When HKCs were co-incubated with TGF-β1 and curcumin for 72 h, curcumin maintained the epithelial morphology in a dose-dependent manner, decreased expression of vimentin, α-SMA and FSP1 normally induced by TGF-β1, and increased expression of E-cadherin, cytokeratin. Importantly, we found that curcumin reduced Akt, mTOR and P70S6K phosphorylation, effectively suppressing the activity of the Akt/mTOR pathway in HKCs. Curcumin also promoted HKC proliferation, and antagonized TGF-β1-driven EMT through the inhibition of Akt/mTOR pathway activity, which may suggest an alternative therapy for renal fibrosis.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2465K) HTML形式で全画面表示 -
Kazuhiro Shima, Masahiro Tsuchiya, Takefumi Oizumi, Teruko Takano-Yama ...2017 年 40 巻 1 号 p. 25-33
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
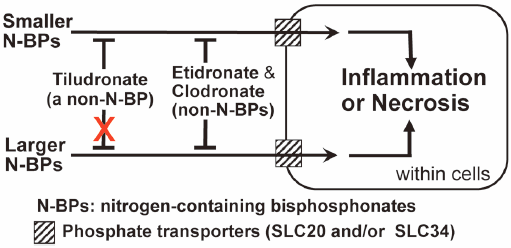
ジャーナル フリー HTMLBisphosphonates (BPs) are used against diseases with enhanced bone resorption. Those classed as nitrogen-containing BPs (N-BPs) exhibit much stronger anti-bone-resorptive effects than non-nitrogen-containing BPs (non-N-BPs). However, N-BPs carry the risk of inflammatory/necrotic side effects. Depending on their side-chains, BPs are divided structurally into cyclic and non-cyclic types. We previously found in mice that etidronate and clodronate (both non-cyclic non-N-BPs) could reduce the inflammatory effects of all three N-BPs tested (cyclic and non-cyclic types), possibly by inhibiting their entry into soft-tissue cells via SLC20 and/or SLC34 phosphate transporters. Tiludronate is the only available cyclic non-N-BP, but its effects on N-BPs’ side effects have not been examined. Here, we compared the effects of etidronate, clodronate, and tiludronate on the inflammatory effects of six N-BPs used in Japan [three cyclic (risedronate, zoledronate, minodronate) and three non-cyclic (pamidronate, alendronate, ibandronate)]. Inflammatory effects were evaluated in mice by measuring the hind-paw-pad swelling induced by subcutaneous injection of an N-BP (either alone or mixed with a non-N-BP) into the hind-paw-pad. All of six N-BPs tested induced inflammation. Etidronate, clodronate, and the SLC20/34 inhibitor phosphonoformate inhibited this inflammation. Tiludronate inhibited the inflammatory effects of all N-BPs except ibandronate and minodronate, which have higher molecular weights than the other N-BPs. The mRNAs of SLC20a1, SLC20a2, and SLC34a2 (but not of SLC34a1 and SLC34a3) were detected in the soft-tissues of hind-paw-pads. These results suggest that etidronate, clodronate, and phosphonoformate may act non-selectively on phosphate transporter members, while tiludronate may not act on those transporting N-BPs of higher molecular weights.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1217K) HTML形式で全画面表示 -
Ryo Hatano, Kotoku Kawaguchi, Fumitaka Togashi, Masato Sugata, Shizuka ...2017 年 40 巻 1 号 p. 34-42
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid that possesses many pharmacological effects, including increasing bile flow, changing the hydrophobicity of the bile acid pool, and modulation of the immune response. UDCA has been approved for treating cholestatic liver disease, such as primary biliary cholangitis. However, several unanticipated severe side effects of UDCA are observed in cholestatic patients, and its pharmacological benefits remain controversial. We reported that ezrin-knockdown (Vil2kd/kd) mice exhibited severe hepatic injury because of a functional disorder in bile duct fluidity and alkalinity regulation, resembling human intrahepatic cholestatic disease. Here we used Vil2kd/kd mice as a cholestatic model to investigate the pharmacological effects of UDCA. We investigated the effects of oral and parenteral administration of UDCA on Vil2kd/kd mice. In Vil2kd/kd mice, fed a 0.5% (w/w) UDCA diet for 3 weeks, hepatic injury was exacerbated, although oral administration of a lower dose of UDCA slightly improved hepatic function in Vil2kd/kd mice. On the other hand, intraperitoneal administration of UDCA (50 mg/kg/d) ameliorated hepatic function and markedly reduced periductal fibrosis and cholangiocyte proliferation in Vil2kd/kd mice although biliary pH and HCO3− concentration were not improved. The expression levels of inflammatory and profibrotic genes were also significantly decreased in these mice. Furthermore, UDCA prevented cholangiocytes from hydrophobic bile acid-induced cytotoxicity independent of extracellular pH in in vitro experiments. These results suggest that an appropriate dosage of UDCA can ameliorate the intrahepatic cholestasis in Vil2kd/kd mice without changing the biliary bicarbonate secretion.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (5469K) HTML形式で全画面表示 -
Hajime Hiyama, Aya Ozawa, Hiroaki Kumazawa, Osami Takeda2017 年 40 巻 1 号 p. 43-48
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe Ephedra herb, which has been used in Kampo medicines, originates from terrestrial stems of Ephedra species. It is important to establish cultivation methods and cultivars to secure a stable supply of the Ephedra herb that would meet the quality standards for the ephedrine alkaloids content. In this study, we first grew Ephedra sinica plants derived from seeds in the field for 5 years. Then, for selective breeding of cultivars that could meet the quality standards for the ephedrine alkaloids content, we measured the content of total alkaloids (TAs), ephedrine (Eph), and pseudoephedrine (PEph) in individual plants derived from seedlings and grown for 4 years in the field. The range of the TA content in each individual plant was narrower than that among individual plants grown in the field. Therefore, individual plants were selected according to their TA content, Eph/PEph ratio, and stolon-formation capability. The selected individuals were propagated using stolons, and their TA content was studied for 2 years. In the second year, the TA content in terrestrial stems derived from stolons of the selected individuals was as high as that of their parents. Therefore, it was confirmed that the selected individuals that were propagated using stolons could produce TA reproducibly. This study suggested that selective breeding using stolon propagation is effective for stabilizing Ephedra herb TA content.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1402K) HTML形式で全画面表示 -
Tatsuki Uemura, Shingo Ito, Yusuke Ohta, Masanori Tachikawa, Takahito ...2017 年 40 巻 1 号 p. 49-55
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLCerebral creatine deficiency syndromes (CCDSs) are caused by loss-of-function mutations in creatine transporter (CRT, SLC6A8), which transports creatine at the blood–brain barrier and into neurons of the central nervous system (CNS). This results in low cerebral creatine levels, and patients exhibit mental retardation, poor language skills and epilepsy. We identified a novel human CRT gene missense mutation (c.1681 G>C, G561R) in Japanese CCDSs patients. The purpose of the present study was to evaluate the reduction of creatine transport in G561R-mutant CRT-expressing 293 cells, and to clarify the mechanism of its functional attenuation. G561R-mutant CRT exhibited greatly reduced creatine transport activity compared to wild-type CRT (WT-CRT) when expressed in 293 cells. Also, the mutant protein is localized mainly in intracellular membrane fraction, while WT-CRT is localized in plasma membrane. Western blot analysis revealed a 68 kDa band of WT-CRT protein in plasma membrane fraction, while G561R-mutant CRT protein predominantly showed bands at 55, 110 and 165 kDa in crude membrane fraction. The bands of both WT-CRT and G561R-mutant CRT were shifted to 50 kDa by N-glycosidase treatment. Our results suggest that the functional impairment of G561R-mutant CRT was probably caused by incomplete N-linked glycosylation due to misfolding during protein maturation, leading to oligomer formation and changes of cellular localization.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (2470K) HTML形式で全画面表示 -
Hirotake Ishida, Shin-ya Saito, Eita Hishinuma, Tomohisa Ishikawa2017 年 40 巻 1 号 p. 56-60
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLHigh K+-induced contraction of arterial smooth muscle is thought to be mediated by membrane depolarization and subsequent activation of voltage-dependent Ca2+ channels (VDCCs). In line with this, this study found that contraction induced by 80 mM K+ was almost abolished by nifedipine (1 µM), a VDCC inhibitor, in isolated rat aorta, and was markedly suppressed in the iliac artery. However, nifedipine (1 µM) only partially suppressed high K+-induced contraction in the tail artery. The contractions remaining in the arteries were further reduced by non-selective cation channel (NSCC) inhibitors, including 2-aminoethoxydiphenyl borate (2-APB) (100 µM), SK&F96365 (10 µM), and 3,4-dihydro-6,7-dimethoxy-α-phenyl-N,N-bis[2-(2,3,4-trimethoxyphenyl)ethyl]-1-isoquinolineacetamide hydrochloride (LOE908) (10 µM). In particular, sustained tonic contraction was nearly abolished. Prazosin (0.3 µM), an α1-adrenoceptor antagonist, partially inhibited high K+-induced contraction in the tail and iliac arteries, but had no effect in the aorta. Consistently, tyramine potently induced contraction in the tail and iliac arteries, but not in the aorta. Furthermore, the inhibition by prazosin and NSCC inhibitors of the high K+-induced contraction in the presence of nifedipine was comparable. These results suggest that depending on the type of artery, high K+-induced contraction is mediated by Ca2+ influx not only through VDCCs but also through NSCCs, the activation of which is due to the activation of α1-adrenoceptors by the released noradrenaline from sympathetic nerve terminals resulting from high K+ stimulation.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (686K) HTML形式で全画面表示 -
Xiaoran Zhao, Bowen Liu, Shui Liu, Lin Wang, Jianfeng Wang2017 年 40 巻 1 号 p. 61-67
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
電子付録Pneumolysin (PLY) is a devastating bacterial protein toxin of Streptococcus pneumoniae that punctures the cytomembrane, leading to pathological reactions, such as cell disruption and inflammation. Drugs capable of closely impacting the toxin are considered advantageous in the treatment of bacterial infections. Amentoflavone (AMF) is a chemical substance extracted from traditional Chinese herbs. Previous studies have demonstrated that AMF has multiple pharmacological effects and mentioned without attenuating pneumolysin-mediated cytotoxicity. This work focuses on the influence of AMF on inhibitory hemolytic mechanisms. AMF interacts with the toxin at Ser254, Glu277, Arg359, and effectively weakens the oligomerization of wild-type PLY and provides considerable protection against pneumolysin-mediated human alveolar epithelial (A549) cell damage. The results of our study demonstrate that AMF could be a candidate against pneumolysin-related injury.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (4457K) HTML形式で全画面表示 -
Hanayo Kodama, Yoshinaga Tamura, Ichiro Kamei, Kyoko Sato, Hiroshi Aki ...2017 年 40 巻 1 号 p. 68-72
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLMicrocrystalline cellulose (MCC) is used globally as an inactive ingredient in food and nutraceutical products and is commonly used as a food additive. To confirm the conformity of MCC to the solubility requirements stipulated in international specifications, the solubilities of commercially available MCC products were tested in sodium hydroxide (NaOH) solution. All of the samples were insoluble in NaOH solution, which is inconsistent with the descriptions provided in international specifications. We also prepared celluloses with different degree of polymerization (DP) values by acid hydrolysis. Celluloses with lower DP were prepared using a three-step process, and their solubilities were tested in NaOH solution. These celluloses were found to be insoluble, which is inconsistent with the descriptions provided in international specifications. The present study suggests that the descriptions of the solubility of the celluloses in NaOH solution found in the current international specifications should be revised.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3721K) HTML形式で全画面表示 -
Yugo Chisaki, Nobuhiko Nakamura, Yoshitaka Yano2017 年 40 巻 1 号 p. 73-81
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe purpose of this study was to propose a time-series modeling and simulation (M&S) strategy for probabilistic cost-effective analysis in cancer chemotherapy using a Monte-Carlo method based on data available from the literature. The simulation included the cost for chemotherapy, for pharmaceutical care for adverse events (AEs) and other medical costs. As an application example, we describe the analysis for the comparison of four regimens, cisplatin plus irinotecan, carboplatin plus paclitaxel, cisplatin plus gemcitabine (GP), and cisplatin plus vinorelbine, for advanced non-small cell lung cancer. The factors, drug efficacy explained by overall survival or time to treatment failure, frequency and severity of AEs, utility value of AEs to determine QOL, the drugs’ and other medical costs in Japan, were included in the model. The simulation was performed and quality adjusted life years (QALY) and incremental cost-effectiveness ratios (ICER) were calculated. An index, percentage of superiority (%SUP) which is the rate of the increased cost vs. QALY-gained plots within the area of positive QALY-gained and also below some threshold values of the ICER, was calculated as functions of threshold values of the ICER. An M&S process was developed, and for the simulation example, the GP regimen was the most cost-effective, in case of threshold values of the ICER=$70000/year, the %SUP for the GP are more than 50%. We developed an M&S process for probabilistic cost-effective analysis, this method would be useful for decision-making in choosing a cancer chemotherapy regimen in terms of pharmacoeconomic.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (814K) HTML形式で全画面表示 -
 Katsuki Nishihashi, Kei Kawashima, Takami Nomura, Yumiko Urakami-Takeb ...2017 年 40 巻 1 号 p. 82-87
Katsuki Nishihashi, Kei Kawashima, Takami Nomura, Yumiko Urakami-Takeb ...2017 年 40 巻 1 号 p. 82-87
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
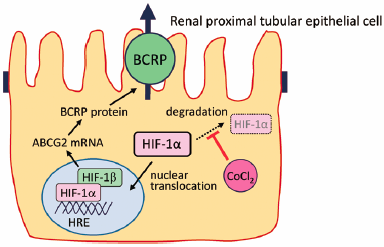
ジャーナル フリー HTMLThe human breast cancer resistance protein (BCRP/ABCG2), a member of the ATP-binding cassette transporter family, is a drug transporter restricting absorption and enhancing excretion of many compounds including anticancer drugs. The cis-regulatory elements in the BCRP promoter include a hypoxia response element, i.e., the DNA binding site for hypoxia-inducible factor-1 (HIF-1). In this study, we investigated the effect of cobalt chloride, a chemical inducer of HIF-1α, on the expression and function of BCRP in human renal proximal tubular cell line HK-2. Cobalt chloride treatment significantly increased the mRNA expression of not only glucose transporter 1 (GLUT1), a typical HIF-1 target gene mRNA, but also ABCG2 mRNA in HK-2 cells. The BCRP inhibitor Ko143-sensitive accumulation of BCRP substrates such as Hoechst33342 and mitoxantrone was significantly enhanced by cobalt chloride treatment. In addition, treatment with cobalt chloride significantly increased the Ko143-sensitive accumulation of fluorescein isothiocyanate-labeled methotrexate in HK-2 cells. Furthermore, cobalt chloride treatment attenuated the cytotoxicity induced by mitoxantrone and methotrexate, which might be, at least in part, due to the increase in BCRP-mediated transport activity via HIF-1 activation. These findings indicate that HIF-1 activation protects renal proximal tubular cells against BCRP substrate-induced cytotoxicity by enhancing the expression and function of BCRP in renal proximal tubular cells.
Graphical Abstract Fullsize Image抄録全体を表示Editor's pickIn their report, Nishihashi et al. describe in detail that cobalt chloride-induced activation of hypoxia-inducible factor-1 (HIF-1) increases not only mRNA and protein expression but also function of breast cancer resistance protein (BCRP/ABCG2) in a human renal proximal tubular epithelial cell line. HIF-1 consists of an inducible a-subunit (HIF-1a) and a constitutive b-subunit (HIF-1b). The stabilization of HIF-1a under hypoxia and various biochemical stimuli leads to nuclear translocation, dimerization with HIF-1b and binding to hypoxia response element (HRE) sequences in the promoter of various target genes. The present results indicate that HIF-1 plays an important role in modulating BCRP-mediated transport activity in renal proximal tubular epithelial cells.
PDF形式でダウンロード (591K) HTML形式で全画面表示 -
Mupeng Li, Yaodong Hu, Huilan Li, Zhipeng Wen, Xiaolei Hu, Daoyu Zhang ...2017 年 40 巻 1 号 p. 88-96
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
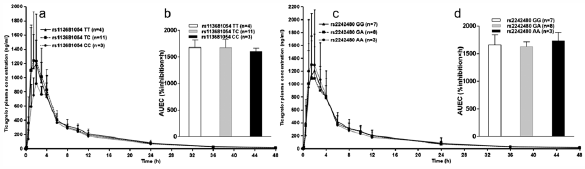
ジャーナル フリー HTMLTicagrelor is a direct-acting P2Y12 receptor antagonist. It is rapidly absorbed and partly metabolized to the active metabolite AR-C124910XX by CYP3A4 and CYP3A5. Three genetic loci (SLCO1B1, CYP3A4, and UGT2B7) were reported to affect ticagrelor pharmacokinetics. This study aimed to investigate the possible effects of SLCO1B1 and CYP3A4/5 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of ticagrelor in healthy Chinese male volunteers. Eighteen healthy male volunteers who participated in pharmacogenetics study of ticagrelor were genotyped for SLCO1B1 rs113681054, SLCO1B1*5 (rs4149056), CYP3A4*1G (rs2242480), and CYP3A5*3 (rs776746). All subjects received a single 180 mg loading dose of ticagrelor and then series blood samples were collected from 0 to 48 h. Plasma concentrations of ticagrelor and AR-C124910XX were determined by the high performance liquid chromatography-tandem mass spectrometry method. Inhibition in platelet aggregation (IPA) was assessed and the area under the time–effect curve (AUEC) for the IPA was calculated as pharmacodynamic parameters. No significant difference in ticagrelor pharmacokinetics among genotypes of the two genes was observed. The AUEC did not differ significantly among genotypes of candidate single nucleotide polymorphisms (SNPs). Our data suggest that common genetic variants in SLCO1B1 and CYP3A4/5 may have no effect on the pharmacokinetics and pharmacodynamics of ticagrelor in healthy Chinese volunteers.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (803K) HTML形式で全画面表示 -
Yoshikazu Sawaguchi, Zuojun Wang2017 年 40 巻 1 号 p. 97-103
発行日: 2017/01/01
公開日: 2017/01/01
[早期公開] 公開日: 2016/10/22 ジャーナル フリー HTML
ジャーナル フリー HTMLRecombinant tissue-type plasminogen activator (rt-PA) is effective and widely used in the treatment of acute ischemic stroke (AIS). However, symptomatic intracranial hemorrhage (ICH), an adverse reaction of rt-PA, is known to occur depending on underlying diseases and rt-PA doses, and to occur more frequently with a greater delay from stroke onset until initiation of rt-PA. Therefore, limitations on the use of rt-PA, such as having to be started within 4.5 h of stroke onset, mean that rt-PA is only indicated in some stroke patients. However, the number of patients in whom rt-PA is indicated could increase if symptomatic ICH induced by rt-PA could be reduced. Therefore, we believe that, if the incidence of adverse reactions such as ICH could be reduced by using lower rt-PA doses together with ultrasound (US), the number of patients eligible for rt-PA treatment would increase. In other words, we hypothesized that, if thrombolysis can be accelerated by US, then recanalization rates similar to currently used doses of rt-PA can be achieved at reduced rt-PA doses. Therefore, to investigate to what extent US enhances the thrombolytic efficacy of rt-PA, the relationship between acceleration of rt-PA thrombolysis and US acoustic intensity was quantitatively evaluated in an in vitro bovine thrombus model. It was found that, within a range of US output that is noninvasive in humans, the combined use of US can increase thrombolytic activity up to 2.5 times more than with rt-PA alone. These findings suggest that US can greatly reduce the required doses of rt-PA.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1366K) HTML形式で全画面表示
-
Nobuaki Matsui, Rie Yoshioka, Asako Nozawa, Naonobu Kobayashi, Yukari ...2017 年 40 巻 1 号 p. 104-107
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLThe contribution of caspases to hepatic ischemia/reperfusion (I/R)-induced apoptosis has not been completely understood yet. Several studies have demonstrated increased caspase activity during I/R and the protective effect of caspase inhibitors against I/R injuries. However, reports with opposing results also exist. Herein, we examined the contribution of caspases to the I/R-induced hepatic apoptosis in rats using caspase inhibitors and specific substrates of caspases. Hepatic I/R was induced via a 2-h occlusion of the portal vein and the hepatic artery, without conducting bile duct occlusion. DNA laddering and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end-labeling (TUNEL)-positive cells were increased at 3 h after reperfusion. Pretreatment with caspase inhibitors (Z-Asp-2,6-dichlorobenzoyloxymethylketone (Z-Asp-cmk) 2 or 10 mg/kg intravenously (i.v.), 20 mg/kg intraperitoneally (i.p.), Z-Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-fmk) 3 mg/kg i.v.) failed to reduce apoptosis induced by I/R. Interestingly, apoptosis induced by the portal triad (hepatic artery, portal vein, and bile duct) occlusion/reperfusion could be marginally attenuated using Z-Asp-cmk (2 mg/kg i.v.). The cleavage activity for Ac-DEVD-α-(4-methylcoumaryl-7-amide) (MCA), a caspase-3/7/8/9 substrate, was significantly increased by I/R. Conversely, the cleavage activities for Ac-DNLD-MCA and MCA-VDQVDGW[K-DNP]-NH2, specific substrates for caspase-3 and -7 respectively, were decreased by I/R. Protein expression of the cellular inhibitor of apoptosis protein 2 (c-IAP2), an endogenous caspase inhibitor, was increased by ischemia. Nuclear translocation of the apoptosis-inducing factor (AIF), an initiator protein of caspase-independent apoptosis, was also increased during I/R. These results suggest that caspases are inhibited by c-IAP2 induced during ischemia and that AIF may be involved in initiation of apoptosis induced by hepatic I/R without bile duct occlusion.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1263K) HTML形式で全画面表示 -
Hitoshi Sasajima, Sadaharu Miyazono, Tomohiro Noguchi, Makoto Kashiway ...2017 年 40 巻 1 号 p. 108-112
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTMLExposure to environmental neurotoxins is suspected to be a risk factor for sporadic progressive neurodegenerative diseases. Parkinson’s disease has been associated with exposure to the pesticide rotenone, a mitochondrial respiration inhibitor. We previously reported that intranasal administration of rotenone in mice induced dopaminergic (DA) neurodegeneration in the olfactory bulb (OB) and reduced olfactory functions. In the present study, we investigated the DA neurons in the brains of mice that were administered rotenone intranasally for an extended period. We found that the olfactory function of mice was attenuated by rotenone administration. Electrophysiological analysis of the mitral cells, which are output neurons in the OB, revealed that the inhibitory input into the mitral cells was retarded. In the immunohistochemical analysis, neurite degeneration of DA neurons in the substantia nigra was observed in rotenone-administered mice, indicating that rotenone progressively initiated the degeneration of cerebral DA neurons via the nasal route.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (3237K) HTML形式で全画面表示 -
Atsunobu Sagara, Takeshi Karasawa, Katsuhide Igarashi, Maky Otsuka, Re ...2017 年 40 巻 1 号 p. 113-117
発行日: 2017/01/01
公開日: 2017/01/01
 ジャーナル フリー HTML
ジャーナル フリー HTML
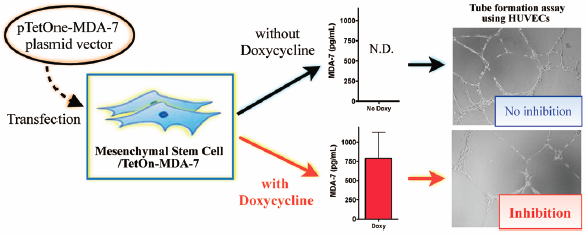
電子付録Mesenchymal stem cells (MSCs) have been explored as a “live” carrier of cytokines for targeted cancer therapy, but, in earlier reports in the literature, the secretion process of therapeutic cytokines was not regulated. The purpose of this study was to generate MSCs to conditionally secrete the melanoma differentiation-associated gene-7 (MDA-7) tumor-suppressor protein. To control the secretion of MDA-7 from MSCs, a well-established tetracycline-controlled transcriptional activation system was incorporated into MDA-7 plasmid. MDA-7 gene expression was induced in the engineered MSCs only in the presence of doxycycline, as characterized by quantitative reverse transcription (qRT)-PCR. Enzyme-linked immunosorbent assay (ELISA) also revealed that the MDA-7 protein was secreted from the engineered MSCs only after the cells had been exposed to doxycycline. Both recombinant human MDA-7 protein and the conditioned medium from the engineered MSCs in the presence of doxycycline significantly inhibited tube formation of human umbilical vascular endothelial cells (HUVECs), indicating that our system could be used for targeted, antiangiogenic therapy. Overall, this study provides useful information on the potential use of engineered MSCs for the controlled secretion of therapeutic proteins, in this case MDA-7, for targeted cancer therapy.
Graphical Abstract Fullsize Image抄録全体を表示PDF形式でダウンロード (1466K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|