
- |<
- <
- 1
- >
- >|
-
Hideyuki KATSUMATAArticle type: Highlights
2019 Volume 35 Issue 12 Pages 1289-1290
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
-
Yaqing ZHANG, Song ZHOU, Xiaoxi CHANG, Xiaoqi TAOArticle type: Rapid Communications
2019 Volume 35 Issue 12 Pages 1291-1293
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: November 22, 2019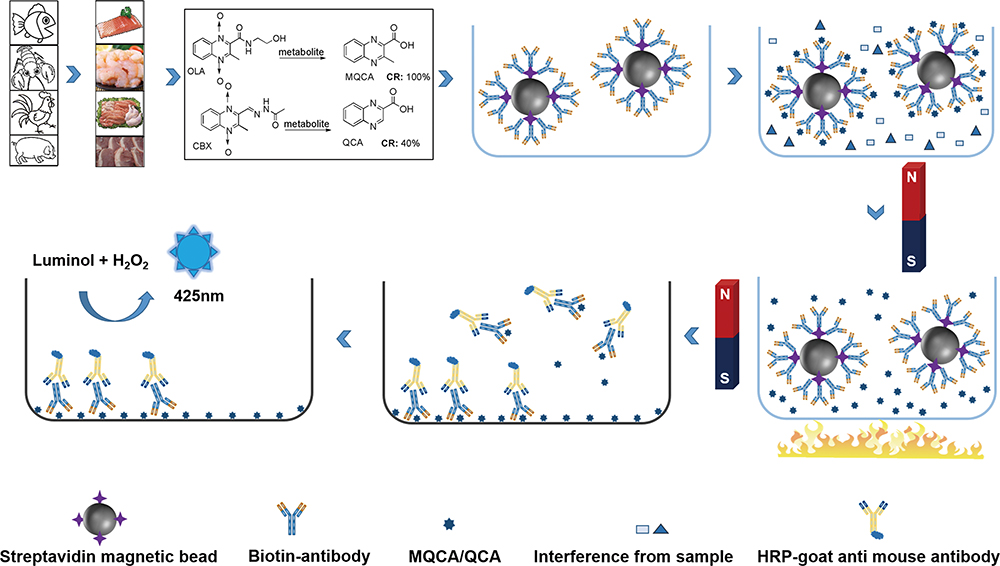 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialWe have reported a sensitive chemiluminescent competitive indirect enzyme-linked immunosorbent assay (CL-ciELISA) based on immunomagnetic beads separation, purification and enrichment for the simultaneous determination of 3-methyl-quinoxaline-2-carboxylic acid (MQCA) and quinoxaline-2-carboxylic acid (QCA) in edible animal tissues. Forty field samples were analyzed with the developed CL-ciELISA, and the results correlated well with those obtained in liquid chromatography–tandem mass spectrometry (LC-MS/MS), confirming the utility of CL-ciELISA for the quantitation of MQCA and QCA in fish, shrimp, pork and chicken with a good accuracy.
View full abstractDownload PDF (2536K) -
Yanbei ZHUArticle type: Rapid Communications
2019 Volume 35 Issue 12 Pages 1295-1298
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: November 22, 2019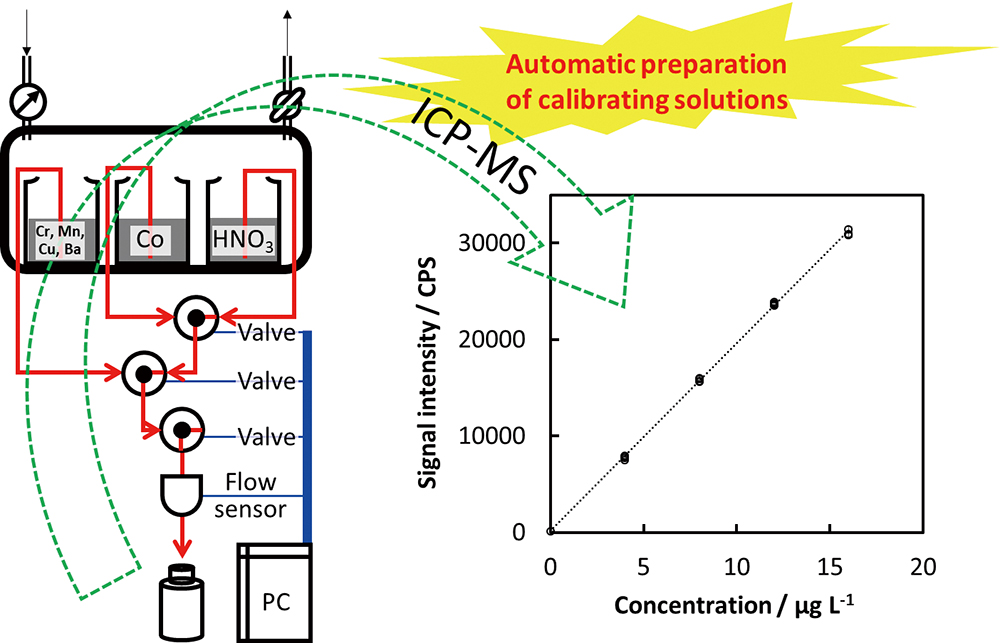 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSAn automatic system was developed to prepare calibrating solutions for elemental measurements by inductively coupled plasma mass spectrometry (ICP-MS). The flow-rates of diluting solution, internal standard solution, and initial elementals solution were monitored with a single ultrasonic flow sensor. Multiple calibrating solutions prepared by the present automatic system were analyzed by ICP-MS, whose results provided a correlation coefficient better than 0.999 for calibration curves. The results for multi-elements in a river-water certified reference material (NMIJ CRM 7202-b) agreed with the certified values.
View full abstractDownload PDF (196K)
-
Xueling SHAN, Xiaomeng SHAN, Tao PAN, Fanzhuo DAI, Xiaohui CHEN, Wench ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1299-1304
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: July 12, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSA solid-state electrochemiluminescence (ECL) sensor for the detection of reduced glutathione (GSH) based on a g-C3N4/SiO2 modified glass carbon electrode (GCE) has been developed in this research. The g-C3N4, which is employed as a luminophore, is simply prepared and exhibits an excellent ECL response. Mesoporous silica hollow spheres (SiO2) with a large specific surface area are introduced here to increase the loading amount of g-C3N4. Compared to a g-C3N4 modified GCE, the g-C3N4/SiO2 modified GCE displays a much higher ECL intensity. A high enhancement effect on the ECL intensity of g-C3N4/SiO2 modified GCE is obtained in the presence of GSH in the electrolyte. Moreover, the enhanced ECL intensity shows a good linear relationship to the GSH concentration in the range from 1.0 × 10−7 to 5.0 × 10−4 M, with a detection limit of 2.0 × 10−8 M (6.1 ng/mL). Besides, the ECL sensor exhibits a good anti-interference ability and has been successfully applied in the detection of GSH in commercial samples. The proposed sensor provides a promising platform for life science.
View full abstractDownload PDF (805K) -
Hanshu WU, Xiaojun YANG, Jinxia MEN, Huibin ZHANG, Jinpei ZHOUArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1305-1310
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: July 12, 2019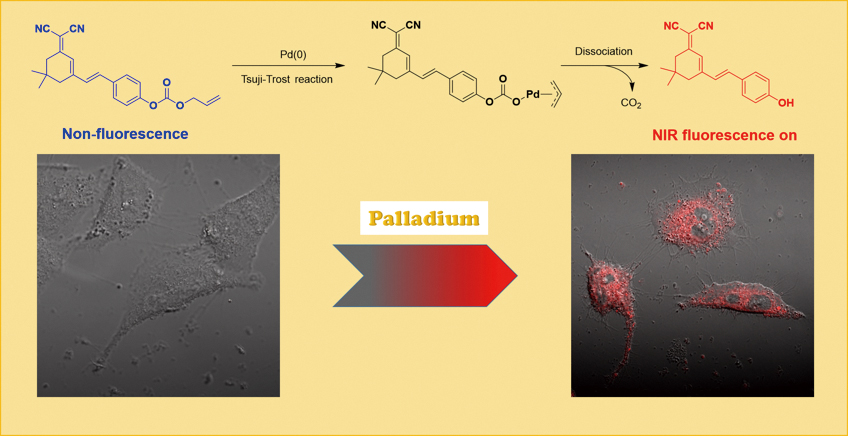 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialPalladium (Pd) has been acknowledged to be a rare inner transition metal, which plays a pivotal role in many fields. This article focuses on developing a safe and effective near-infrared fluorescent probe, MW-PD, which would make a great contribution to the detection of palladium residue in drugs, especially trace residues. The fluorescent probe was rationally designed by combining the dicyanoisophorone fluorophore with an allyloxycarbonyl group. Based on the Tsuji–Trost reaction, the probe exhibited high selectivity and sensitivity toward Pd (0) over other common metal ions with a low detection limit (8.0 nM). Moreover, MW-PD showed biocompatibility and was successfully applied to imaging Pd (0) in Hela cells.
View full abstractDownload PDF (839K) -
Miki ISODA, Makoto FUKUMA, Akira HARATAArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1311-1315
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialThe first demonstration of a photothermal heterodyne interferometer (PHI) combined with micro-HPLC (high-performance liquid chromatography) is reported. A semiconductor laser (375 nm) was used for excitation, and the temperature change caused by heat released from photoexcited species was detected with a He–Ne laser (632.8 nm). The temperature-dependent refractive index change of the solvent modified the optical path of the probe beam. The phase difference between two arms of the interferometer, one passing through the heated sample and another as a reference, was sensitively detected with the PHI. The nitro-polycyclic aromatic hydrocarbon and vitamin mixture separated via micro-HPLC was successfully detected with the PHI as well as a UV detector. The detection limit of the PHI for riboflavin in the absorbance units was 77 times better than that of the commercial UV detector. The detection limit of the PHI with a small flow cell (6 nL) was the same as that with a large flow cell (18 nL) for 1-nitropyrene.
View full abstractDownload PDF (337K) -
Zhimin LUO, Xuan LI, Lu WANG, Chun CHANG, Qiang FUArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1317-1325
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 09, 2019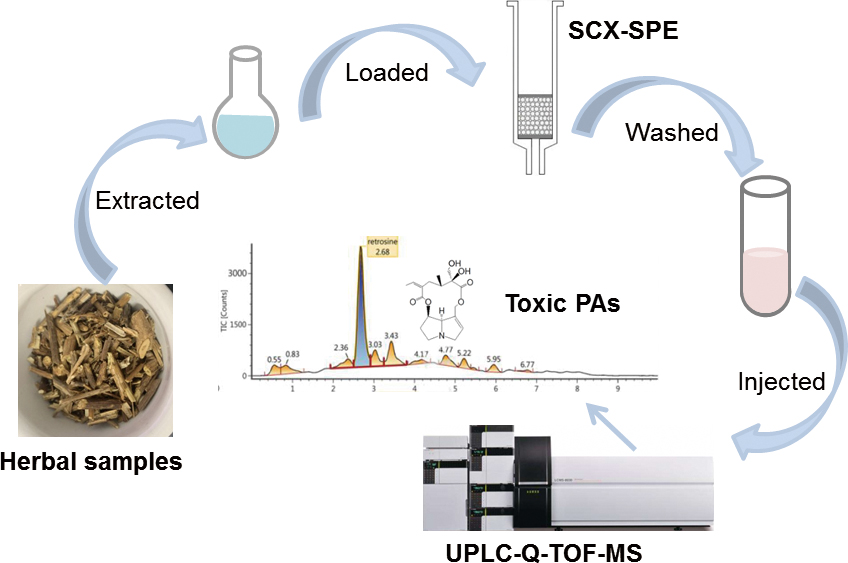 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSPyrrolizidine alkaloids are secondary metabolites of plants and can cause significant hepatotoxicity in humans. In this study, a fast and simple method was developed to determine ten pyrrolizidine alkaloids (PAs) in six types of herbal medicines using ultra performance liquid chromatography–quadrupole–time of flight mass spectrometry (UPLC-Q-TOF-MS). An efficient solid-phase extraction procedure was carried out by using strong cation-exchange cartridges and the parameters were optimized. The established analytical method was validated and the results showed that the method presented satisfactory accuracy and precision. The established method was successfully applied for the determination of PAs in six herbal plants, including Senecionis Scandentis Hebra, Arnebiae Radix, Asteris Radix Et Rhizoma, Farfarae Flos, Senecionis Cannabifolii Herba and Emilia sonchifolia. PAs were found in all of these herbal plant samples. Eight types of related commercial herbal drugs were also detected, six of them were detected with different amounts of PAs. This work not only provided a powerful technical platform for both qualitative and quantitative analysis of PAs in herbal medicines, but also obtained information concerning PAs in these herbal samples, which could provide reference to the government regulatory authorities and non-governmental organizations.
View full abstractDownload PDF (2258K) -
Takuya YONESAKA, Souta YAGI, Takashi YASUI, Akio YUCHIArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1327-1331
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 09, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialIon exchanges by aromatic anions (AAs) of varying volumes on anion exchange resins of different cross linking (CL)-degrees were studied. The exchanges on the resins with low CL-degrees having the larger and the less independent exchange sites proceeded fast and completely at a high AA concentration; the selectivity coefficient increased with the progress of the exchange, due to the cooperative interaction between exchanged AAs. In contrast, in the exchanges on the resins with a CL-degree of 8% having the smaller (more specifically, having the average volume of 300 Å3 with a variance) and the more independent exchange sites, the large AA may not enter the exchange sites having sizes smaller than the volume of AA to cause mid-saturation of ion exchange and to increase the reaction time because of the need for detouring; the selectivity coefficients were substantially independent of the progress of the exchange, due to the isolation of each AA.
View full abstractDownload PDF (161K) -
Yuki YAGI, Akira OKAZAKI, Megumi ENDO, Kumi YANAGISAWA, Jun FUKUDA, Ko ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1333-1340
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 16, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSHuman antithrombin (AT) has two isoforms of which the predominant α-form is glycosylated on all four possible glycosylation sites and the lower abundant β-isoform lacks the oligosaccharide on Asn135. The main oligosaccharide structure of human AT consists of biantennary complex-type oligosaccharides lacking a core fucose. Generally, Chinese hamster ovary (CHO) cells produce recombinant human AT (rhAT) with core-fucosylated oligosaccharides. However, rhAT lacking core-fucose oligosaccharides can be produced by POTELLIGENT® technology, which uses FUT8 knockout CHO cells in production. The rhAT has more variable glycan structures, such as tetra-antennary complex type, high-mannose type, and mannose 6-phosphate species as minor components compared to plasma-derived human AT (phAT). In addition, the site-specific glycan profile was different between two ATs. We evaluated the effect of these properties on efficacy and safety based on a comparison of rhAT made by that technology with phAT in terms of their respective oligosaccharide structures, site-specific oligosaccharide profiles, and the ratio of α- and β-forms. Although some structural differences were found between the rhAT and phAT, we concluded that these differences have no significant effect on the efficacy and safety of rhAT.
View full abstractDownload PDF (703K) -
Wenzhi XU, Xue LI, Jiwei YIN, Weiyan LIU, Yutao YANG, Wei LIArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1341-1345
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialHydrazine is an important catalyst and chemical raw material. But it is highly toxic and potentially carcinogenic. We designed a new hydrazine probe based on a synergistic effect by introducing acetate and phthalimide into 2-phenyl-benzimidazole (PBI). Comparative experiments proved that “the dual position interaction” had a “synergistic effect” on fluorescence enhancement. The fluorescence enhancement caused by the probe (15.0 fold) is much larger than the sum of the fluorescence enhancement of the two monomer compounds (2.6 and 1.4 folds, respectively). A theoretical calculation showed an inhibition of the PET process and a recovery of the ICT process led to a fluorescence enhancement. The probe was specific to hydrazine and showed a linear response to it in the concentrations range of 0.2 – 200 μM with a LOD of 0.062 μM (1.99 ppb). Moreover, the probe could detect hydrazine in tap water; the recovery of hydrazine from the tap water was between 98.86 – 103.28%.
View full abstractDownload PDF (788K) -
Xue CHU, Yasuro FUSE, Takato SASAKI, Ichiro AIZAWA, Masahiro OGUCHI, Y ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1347-1352
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 16, 2019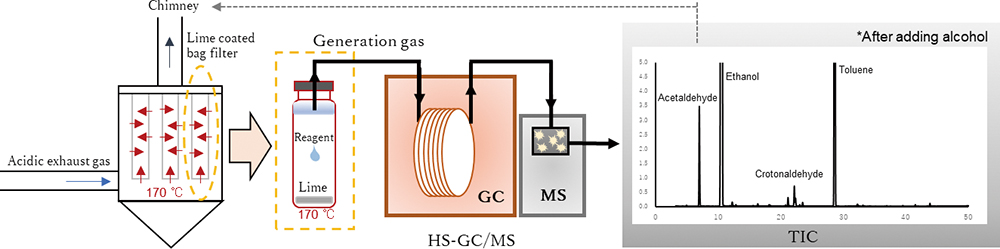 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSAcid gases generated during the thermal treatment of waste are neutralized using devices, such as bag filters coated with slaked lime. However, residual trace organic substances can react with the highly reactive slaked lime. This study investigates the dynamics of organic substances generated in the bag filter when slaked lime is used in the exhaust gas treatment process. The mechanism of aldehyde generation was clarified using head space gas chromatography mass spectrometer (HS-GC/MS). Results indicated that methanol was converted to formaldehyde at a conversion ratio of 0.097% and ethanol was converted to acetaldehyde at a conversion ratio of 0.260%. In addition, when amines used as emulsifiers during slaked lime production persisted in the matrix, acetaldehyde formed at a maximum concentration of 121 mg/m3. The simulation method developed in this study can be used for the initial evaluation of aldehydes unexpectedly produced in an incineration treatment facility.
View full abstractDownload PDF (462K) -
Tomoya SUZUKI, Takeshi OGATA, Mikiya TANAKA, Tohru KOBAYASHI, Hideaki ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1353-1360
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 23, 2019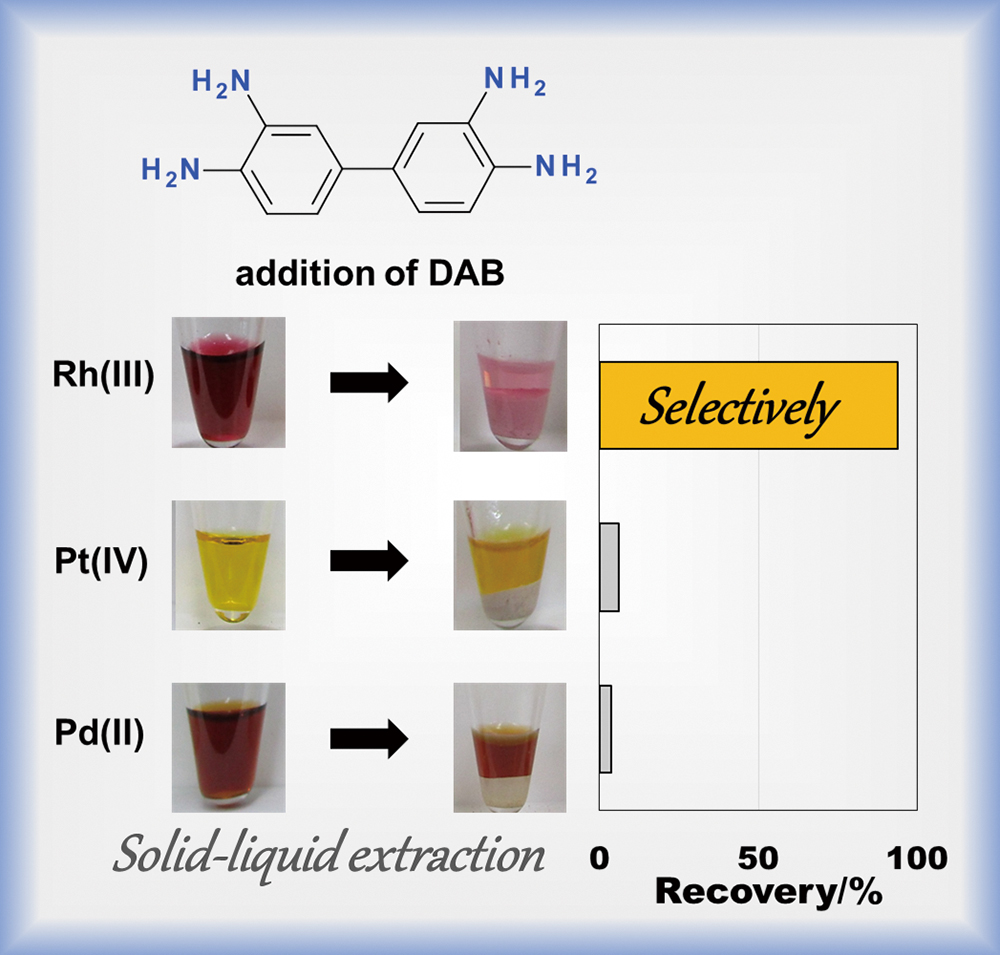 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialThe effective recovery of Rh(III) from mixtures also containing Pd(II) and Pt(IV) is one of the most difficult tasks in platinum group metal refining. Adding 3,3′-diaminobenzidine (DAB) to 7 and 10 M HCl aqueous solutions containing Rh(III), Pd(II), and Pt(IV) chlorido species affords the effective separation of Rh(III) from Pd(II) and Pt(IV) through a process where Rh(III) becomes sequestered into solid phases composed of DAB. The stoichiometry and inner coordination sphere of the metal in Rh–DAB complexes were determined by estimating the Rh(III), H+, and Cl− concentrations in the solid phase and X-ray absorption fine structure measurements to clarify the mechanism of DAB selectivity for Rh(III). These results indicate that the Rh–DAB reaction in a concentrated HCl solution occurs in two steps: (1) the precipitation of DAB trihydrochloride salts, where DAB’s amino groups are protonated and (2) anion exchange of the trihydrochloride salts for chloride ions with [RhCl6]3−, which is the predominant species in a concentrated HCl solution. By contrast, ion-pair complexes with [PdCl4]2− and [PtCl6]2− were not observed in DAB phases. The significantly lower affinity of the DAB trihydro cation for [PtCl6]2− and [PdCl4]2− than for [RhCl6]3− in 7 and 10 M HCl solutions accounts for the effective separation of Rh(III) from Pd(II) and Pt(IV).
View full abstractDownload PDF (739K) -
Masafumi IWATA, Tomohiro UCHIMURAArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1361-1365
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 23, 2019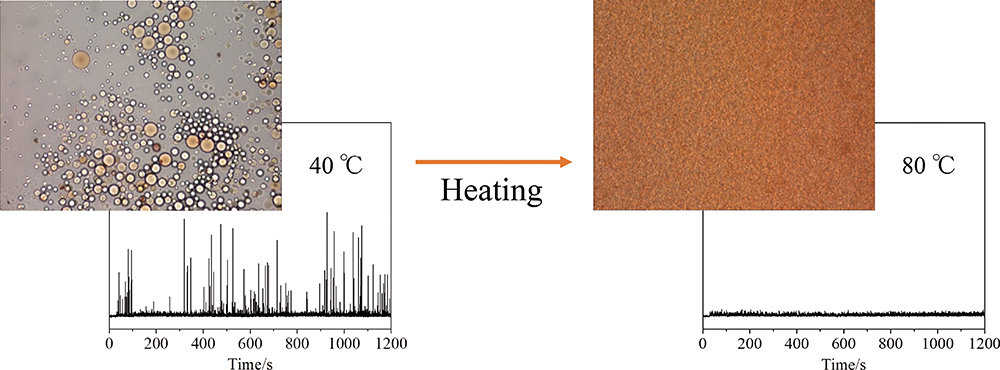 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSThis study used resonance-enhanced multiphoton ionization time-of-flight mass spectrometry (REMPI-TOFMS) to evaluate the phase inversion that is driven via temperature change. A change in temperature prompts phase inversion in an oil-in-water (O/W) emulsion that uses a nonionic surfactant as an emulsifier. In this study, an O/W emulsion was prepared containing Triton X-100 as an emulsifier. To promote emulsion inversion, we heated only the outlet of a capillary column that was used for sample introduction during REMPI-TOFMS. When the emulsion was continuously measured at 40°C, several intense spikes could be detected on a time profile for the analyte, toluene, constructed by extracting the peak areas from a series of obtained mass spectra. This indicated the presence of toluene droplets in the O/W emulsion. No intense spikes appeared at 80°C, however, which suggested the shrinkage or even disappearance of the oil droplets following phase inversion. Using this technique, an emulsion can be measured without affecting the influence of vaporization even at elevated temperatures, which surely is a serious concern when using other analytical techniques. Therefore, this technique would be quite useful for evaluating the phase inversion of an emulsion at elevated temperatures.
View full abstractDownload PDF (282K) -
Jin-Sung PARK, Geunseob OH, Jiwon KIM, Eun Young PARK, Jennifer H. SHI ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1367-1373
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 30, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialC. elegans exhibits a directional migration toward a remembered temperature setpoint (Ts) by activating thermo-sensorimotor neurons. While cryophilic thermotaxis is well reproduced, thermophilic thermotaxis requires very stringent temperature regulations – otherwise, worms exhibit random migration in colder side of Ts. Here, we introduce a thermal stimulus device developed to control worms with different thermotactic behaviors on both colder and warmer sides of the Ts. On a linear gradient, the worm population displayed a Gaussian distribution near Ts but in a skewed shape with a peak shifted to the colder side due to their atactic motion in colder temperature than Ts. By repetitive application of thermal gradient-reversals, we found that their population density became higher near Ts because the speed at which the worms accumulate toward Ts was much faster than that of the dispersion by diffusion to the cold side, resulting in forced aggregation of worms at the desired temperature.
View full abstractDownload PDF (5696K) -
Tetsuo INUI, Hamana SHIROTA, Shoji SAKAOArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1375-1379
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 30, 2019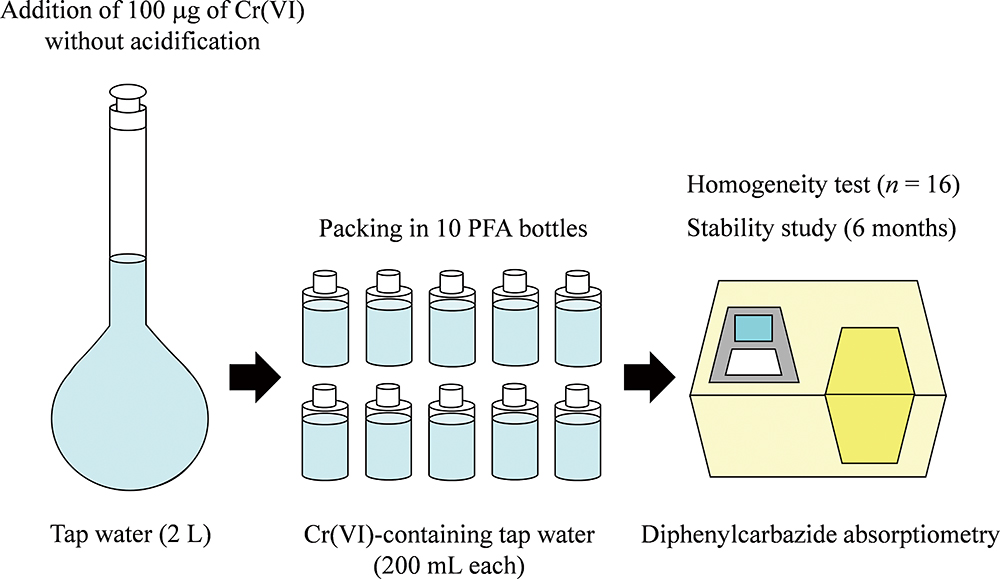 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSWe developed a reference material (RM) for the determination of hexavalent chromium (Cr(VI)) in tap water. The tap water RM was prepared by adding a Cr(VI) standard solution to the raw material without acidification, i.e., under the original pH conditions of 7.6, because the decrease in the concentration of Cr(VI) was observed when the tap water had been adjusted to pH 1 with HNO3. The prepared tap-water RM (2 L) was packed in 10 fluororesin (PFA) bottles with an inside plug (200 mL each). Each PFA bottle (Cr(VI)-containing tap water) was sealed in a reclosable poly bag and then stored at 5°C in a refrigerator. The tap water RM had a Cr(VI) concentration of 51 μg L−1. The concentration of Cr(VI) was determined by diphenylcarbazide absorptiometry using a 100-mm quartz cell. The detection limit of Cr(VI) in the sample solution corresponding to three-times the standard deviation (n = 5) of blank values was 0.51 μg L−1. The homogeneity of Cr(VI) in the tap water RM was evaluated by an analysis of the variance after the Cochran test. There was no significant difference between the within-bottle and between-bottle variances of the analytical results, indicating that the tap water RM was sufficiently homogeneous. The stability of Cr(VI) in the tap water RM was investigated by monitoring the Cr(VI) concentration over a period of 6 months. The slope of the regression line of the Cr(VI) concentration versus the storage time did not significantly differ from zero, indicating that the tap water RM was stable for 6 months. The concentrations (50 – 51 μg L−1) of Cr(VI) in the tap water RM were in good agreement with the total chromium concentrations (50 – 51 μg L−1) obtained by atomic absorption spectrometry.
View full abstractDownload PDF (118K) -
Xinlong ZHOU, Yi CHEN, Qing YANG, Yunong LIU, Yuchen WU, Rongsheng LU, ...Article type: Original Papers
2019 Volume 35 Issue 12 Pages 1381-1384
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: September 13, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSTo improve the accuracy of total polar compounds (TPC) quantification in frying oils by low-field nuclear magnetic resonance (LF-NMR), an optimized statistical method was proposed. The method uses a specially designed sequence to detect the NMR signal in frying oils, and establishes the TPC prediction model by partial least squares (PLS) regression on relaxation properties extracted from the NMR signal. Compared with inversion recovery (IR) and Carr–Purcell–Meiboom–Gill (CPMG) sequences, the designed sequence provides more relaxation information. The experimental result shows that the proposed method is more accurate than reported methods that are based on longitudinal and transverse relaxation times in the TPC quantification of frying oils.
View full abstractDownload PDF (413K) -
Asmaa Kamal EL-DEEN, Kuniyoshi SHIMIZUArticle type: Original Papers
2019 Volume 35 Issue 12 Pages 1385-1391
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
 JOURNAL FREE ACCESS
JOURNAL FREE ACCESS
Supplementary materialThis study deals with the application of different monoterpenes as relatively green bio-based solvents for low density-dispersive liquid–liquid microextraction (LD-DLLME) of different non-steroidal anti-inflammatory drugs (NSAIDs) from aqueous samples in comparison with conventional halogenated solvents. Results indicated that D-limonene could extract the hydrophobic compounds with higher %recovery compared with other bio-based and halogenated solvents. The LD-DLLME procedure was optimized by applying one-factor-at-a-time (OFAT) followed by central composite face-centered design (CCF) of the response surface methodology (RSM). Under the optimal conditions, the method exhibited good linearity with r ≥ 0.9950 with low LOD as well as LOQ levels in the range of 0.11 to 0.81 and 0.36 to 2.69 ng/mL, respectively. The method was efficiently applied to a variety of real aqueous samples. Finally, the green aspect of our procedure was compared with some reported methods using two green metric tools.
View full abstractDownload PDF (362K)
-
Yukihiro ESAKA, Saki KUNISHIMA, Hiromitsu ARUGA, Takuhei YAMAMOTO, Hir ...Article type: Notes
2019 Volume 35 Issue 12 Pages 1393-1397
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
Advance online publication: August 30, 2019 JOURNAL FREE ACCESS
JOURNAL FREE ACCESSCyclic-1,N2-propano-2′-deoxyguanosine-d7 (CPr-dG-d7) was prepared as an isotopic internal standard (IS) for electrospray ionization tandem mass spectrometry (ESI-MS/MS) quantification of CPr-dG in DNA as a candidate cancer risk marker of acetaldehyde intake, mainly from drinking. The deuterated compound was reasonably synthesized from acetaldehyde-d4 and 2′-deoxyguanosine in deuterium oxide (D2O), preventing the deuterium atoms of acetaldehyde-d4 from being substituted by hydrogen atoms, which occurred seriously in aqueous synthesis media via keto–enol tautomerism. Furthermore, another deuterium atom was added from D2O to form CPr-dG-d7. After four weeks of storage in H2O at 10°C, CPr-dG-d7 was found to be sufficiently stable for practical use. The calibration curve of CPr-dG by using a hydrophilic interaction chromatography–ESI-MS/MS system with CPr-dG-d7 as the IS showed sufficient linearity from 1.0 × 10−10 to 4.0 × 10−9 M with r2 = 0.998.
View full abstractDownload PDF (303K)
-
Article type: Announcements
2019 Volume 35 Issue 12 Pages 1399
Published: December 10, 2019
Released on J-STAGE: December 10, 2019
JOURNAL FREE ACCESSDownload PDF (1165K)
- |<
- <
- 1
- >
- >|