
- |<
- <
- 1
- >
- >|
-
Takahiro Mori2023 Volume 71 Issue 3 Pages 188-197
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
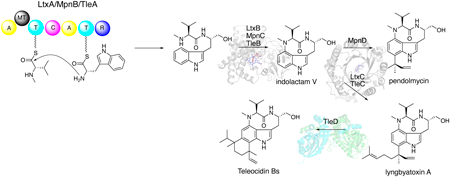
JOURNAL FREE ACCESS FULL-TEXT HTMLTeleocidins are natural products belonging to the indole alkaloid family and show potent protein kinase C activation activity. The structural feature of teleocidins is an indole-fused nine-membered lactam ring structure. Due to their unique structures and strong biological activities, many total synthesis and biosynthetic studies of teleocidins have been performed. Teleocidin biosynthesis involves interesting enzymatic reactions that are challenging in organic synthesis, including oxidative intramolecular C–N bond-forming reactions, regio- and stereo-selective reverse prenylation reactions, and methylation-triggered terpene cyclization. This review summarizes the recent research on functional and structural analyses, as well as enzyme engineering, of teleocidin biosynthetic enzymes.
View full abstractDownload PDF (5875K) Full view HTML
-
Jun Kawahara, Miyako Yoshida, Honami Kojima, Rio Uno, Minoru Ozeki, Ik ...2023 Volume 71 Issue 3 Pages 198-205
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLThe purpose of the present study was to evaluate bitterness suppression effect of adenylic acid (AMP) as a nucleotide-derived nutrient enhancer on a bitter commercial drug. In the present study, we evaluated peripheral bitterness inhibition effect of AMP on the trimethoprim (TMP) and sulfamethoxazole (SMZ) combination formulation based on taste sensor. The taste sensor values of TMP solutions with different concentrations show large sensor output in correlation with the concentration of TMP, whereas no sensor output in shown for the SMZ solutions. Therefore, the bitterness of this combination formulation is mainly due to TMP. We evaluated the TMP bitterness inhibitory effects of AMP, sodium salt of AMP (AMP Na; sodium adenylate), sodium salt of GMP (GMP Na; sodium guanylate), and sodium salt of inosine monophosphate (IMP Na; sodium inosinate), and found that only AMP displayed very effective bitterness inhibition. MarvinSketch analysis revealed that potential electrostatic interaction between cationized TMP and anionized forms (II and III) of AMP may cause bitterness suppression. 1H-NMR study suggested an interaction of TMP and AMP molecules based on chemical shift perturbations and an interaction between the phosphate group of AMP and amino group of TMP. Lastly, conventional elution analysis simulating oral cavity capacity for up to one minute were performed using commercial TMP/SMZ combination granules. The sensor output gradually increased up to 60 s. The addition of AMP solution to the eluted sample at 60 s significantly decreased the bitterness sensor output of the eluted sample.
View full abstractDownload PDF (1759K) Full view HTML -
Liang Xing, Guoliang Gong, Xinyang Chen, Xin Chen2023 Volume 71 Issue 3 Pages 206-212
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
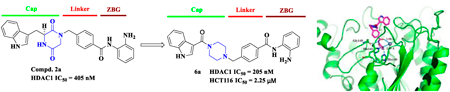
Supplementary materialHistone deacetylases (HDACs) are important targets in cancer treatment, and the development of selective and broad-spectrum HDACs inhibitors (HDACis) is urgent. In this research, a series of aroylpiperazine hybrid derivatives were designed and synthesized. Among these, indole-piperazine hybrids 6a (IC50 = 205 nM) and 6b (IC50 = 280 nM) showed submicromolar activity against HDAC1. Moreover, 6a showed a preferable affinity toward class I HDACs, especially for HDAC1–3. In vitro, 6a exhibited better antiproliferative activities against K562 and HCT116 cell lines than chidamide.
View full abstractDownload PDF (3908K) Full view HTML -
 Asami Ono, Rena Kurihara, Katsuhide Terada, Kiyohiko Sugano2023 Volume 71 Issue 3 Pages 213-219
Asami Ono, Rena Kurihara, Katsuhide Terada, Kiyohiko Sugano2023 Volume 71 Issue 3 Pages 213-219
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLThe purpose of the present study was to provide the experimental and theoretical basis of bioequivalence (BE) dissolution test criteria for formulation development of high solubility-low permeability drugs. According to the biowaiver scheme based on the biopharmaceutics classification system (BCS), for BCS class III drugs, a test formulation and a reference formulation are predicted to be BE when 85% of the drug dissolves within 15 min (T85% < 15 min) in the compendial dissolution test. However, previous theoretical simulation studies have suggested that this criterion may possibly be relaxed for use in practical formulation development. In the present study, the dissolution profiles of 14 famotidine formulations for which BE has been clinically confirmed were evaluated by the compendial dissolution test at pH 1.2 and 6.8. The plasma concentration–time profiles of famotidine formulations were simulated using the dissolution data. In addition, virtual simulations were performed to estimate the range of dissolution rates to be bioequivalent. The fastest and slowest dissolution rates among the famotidine formulations were T85% = 10 min and T85% = 60 min at pH 6.8, respectively. The virtual simulation BE study suggested that famotidine formulations can be bioequivalent when T85% < 99 min. In the case of BCS III drugs, the rate-limiting step of oral drug absorption is the membrane permeation process rather than the dissolution process. Therefore, a difference in the dissolution process has less effect on BE. These results contribute to a better understanding of the biowaiver approach and would be of great help in the formulation development of BCS class III drugs.
View full abstractEditor's pickThe biowaiver scheme based on the biopharmaceutics classification system (BCS-BWS) is used not only as terms of regulatory submissions but also as an indicator for formulation development in drug discovery. The authors investigated the in vitro dissolution rates of formulations of a BCS class III drug and compared them with the criterion in the BCS-BWS. They also discussed the impact of dissolution rates on bioequivalence for BCS class III drugs by virtual simulation. These findings contribute to a better understanding of the biowaiver approach and would help researchers in the formulation development of BCS class III drugs.
Download PDF (1118K) Full view HTML -
Tian Li, Yuxiao Hu, Baojun Shi, Wenjun Wu2023 Volume 71 Issue 3 Pages 220-228
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
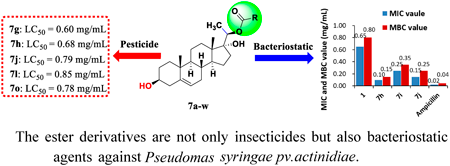
Supplementary materialThere is no doubt that derivation of intermediates from natural product is a very efficient way to develop new environmentally friendly pesticide. We synthesis a succession of compounds esterified with pregn-5-ene-3β,17α,20(S)-triol to evaluate its insecticidal and bacteriostatic activity. Otherwise, their structure–activity relationships (SAR) are also discussed. As a result, compounds 7g, 7h, 7j, 7l and 7o exhibit more obvious insecticidal activity against 3rd Mythimna separata Walker (LC50 = 0.60, 0.68, 0.79, 0.85 and 0.78 mg/mL, respectively) than periplocoside F (PSF). Meanwhile, compounds 7g, 7h and 7i perform well inhibitory activity against Pseudomas syringae pv. actinidiae (Psa) in vitro (minimum inhibitory concentration (MIC) values: 0.10–0.25 mg/mL, minimum bactericidal concentration (MBC) values: 0.15–0.35 mg/mL). And SAR analysis indicates that the replacement and position of fluorine atom on benzoyl are highly vital to biological activity.
View full abstractDownload PDF (1471K) Full view HTML -
Shinya Kimura, Tomoki Komiyama, Tatsuki Masuzawa, Masashi Yokoya, Taka ...2023 Volume 71 Issue 3 Pages 229-233
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialIn vitro evaluation of the physical properties of biopolymer-based hydrogels can help in understanding certain phenomena, such as liquid–liquid phase separation. The formation of bovine serum albumin (BSA) hydrogels was investigated in the pH range of 1.0 to 4.0. Hydrogels were formed in the pH range of 3.0 to 4.0, whereas viscous solutions were formed in the pH range of 1.5 to 2.5. Unexpectedly, formation of BSA hydrogel was observed in extremely acidic condition (pH 1.0). The circular dichroism spectra of BSA solutions were recorded at pH values of 1.0, 2.0, 3.0, and 7.0, and α-helix contents were determined from the ellipticity data at 222 nm. The α-helix content decreased with a decrease in pH, and this decrease was associated with the partial denaturation (F-isoform) and the denaturation (E-isoform) of BSA. However, the α-helix contents at pH 1.0 and 3.0 were similar. BSA hydrogels at pH 1.0 and 3.5 showed similar dynamic viscoelastic properties, further supporting the stereo structural change of BSA from the denatured E-isoform to the partially denatured F-isoform at pH 1.0. The study also focused on measuring viscoelasticity, a fundamental physical property of hydrogels, using traditional rheometer and with minimal sample volume. A highly reproducible procedure for measuring the viscoelastic properties of hydrogels was established using sample volumes of 200 and 350 μL.
View full abstractDownload PDF (2812K) Full view HTML -
Hiroyuki Watanabe, Takuji Ide, Masahiro Ono2023 Volume 71 Issue 3 Pages 234-239
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLIt is generally accepted that the orexin 2 receptor (OX2R) plays a critical role in the arousal-promoting function, and in vivo imaging of OX2R is expected to contribute to elucidation of orexin systems and the development of drugs to treat sleep disorder. In this study, we newly synthesized and characterized a radioiodinated triazole-pyrolidine derivative ([125I]TPI) to detect OX2R in the brain. In vitro studies using OX1R or OX2R expression cells showed selective binding of [125I]TPI to OX2R. In addition, in vitro autoradiography using rat brain sections showed high accumulation of radioactivity in the OX2R expression region. However, [125I]TPI showed low brain uptake in normal mice. These results suggest that [125I]TPI has a fundamental character to detect OX2R in vitro, but further structural modification to improve brain pharmacokinetics is required to use it for in vivo detection of OX2R.
View full abstractEditor's pickThe orexin 2 receptor plays a critical role in the arousal-promoting function. In vivo imaging of orexin 2 receptor is expected to contribute to elucidation of orexin systems and the development of drugs to treat sleep disorder. The authors newly developed a radioiodinated triazole-pyrolidine derivative to detect orexin 2 receptor in the brain. The authors described that an additional structure-activity relationship study based on the triazole-pyrolidine scaffold to improve brain pharmacokinetics may lead to the development of useful orexin 2 receptor imaging probes.
Download PDF (948K) Full view HTML -
Yoshiaki Kitamura, Mahmoud Kandeel, Erina Oba, Chiori Iwai, Keitaro Ir ...2023 Volume 71 Issue 3 Pages 240-249
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
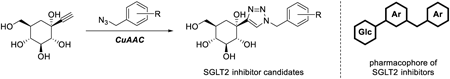
Supplementary materialSodium-glucose cotransporter 2 (SGLT2) inhibitors are clinically available to control blood glucose levels in diabetic patients via an insulin-independent mechanism. It was found that some carbasugar analogs of known SGLT2 inhibitors exert a high inhibiting ability toward SGLT2 and have a prolonged blood glucose lowering effect. In this study, we designed new candidates of carbasugar SGLT2 inhibitor that can be synthesized using copper-catalyzed azide–alkyne cycloaddition (CuAAC) into an aromatic ring, which is a part of the pharmacophore at the final stage in the synthetic protocol for the easier discovery of superior SGLT2 inhibitors. Based on the results of molecular docking studies, some selected compounds have been synthesized. Evaluation of these compounds using a cell-based assay revealed that the majority of these compounds had SGLT2 inhibitory activity in a dose-dependent manner. The SGLT2 inhibitory activity of 7b and 7c was almost equal to that of SGLT2 inhibitors in current use. Furthermore, molecular dynamics simulations also revealed that 7c is a promising novel SGLT2 inhibitor.
View full abstractDownload PDF (1839K) Full view HTML -
Makoto Oba, Mika Shibuya, Yuto Yamaberi, Hidetomo Yokoo, Satoshi Uchid ...2023 Volume 71 Issue 3 Pages 250-256
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialAmphipathic peptides composed of cationic amino acids and hydrophobic amino acids have cell-penetrating ability and are often used as a delivery tool for membrane-impermeable compounds. Small interfering RNA (siRNAs) are one of the delivery targets for such cell-penetrating peptides (CPPs). Cationic CPPs can associate with anionic siRNAs by electrostatic interactions resulting in the formation of nano-sized complexes, which can deliver siRNAs intracellularly. CPPs containing unnatural amino acids offer promising tools to siRNA delivery. However, the detailed structure–activity relationship in siRNA delivery has been rarely studied. In the current study, we designed peptides containing dipropylglycine (Dpg) and explored the cellular uptake and cytotoxicity of peptide/siRNA complexes. The amphipathic structure of the peptides played a key role in complexation with siRNAs and intracellular siRNA delivery. In the amphipathic peptides, cellular uptake of siRNA increased with increasing peptide length, but cytotoxicity was reduced. A peptide containing four Dpg exhibited an effective gene-silencing effect with small amounts of peptides without cytotoxicity in medium containing serum. These findings will be helpful for the design of novel CPPs for siRNA delivery.
View full abstractEditor's pickCell-penetrating peptides (CPPs) are promising intracellular delivery tools for membrane-impermeable compounds such as small interfering RNAs (siRNAs). In this study, the authors designed amphipathic CPPs containing unnatural amino acids dipropylglycine (Dpg) and explored the cellular uptake and cytotoxicity of peptide/siRNA complexes. The results suggested that the amphipathic structure of peptides played a key role in complexation with siRNAs and intracellular siRNA delivery. A Dpg-containing peptide formed an amphipathic a-helical structure and achieved effective intracellular delivery using small amounts of peptides with negligible cytotoxicity. These findings could be valuable for the design of novel CPPs for siRNA delivery.
Download PDF (3187K) Full view HTML
-
Yuta Hatanaka, Hiromasa Uchiyama, Shingo Furukawa, Mai Takase, Shinya ...2023 Volume 71 Issue 3 Pages 257-261
Published: March 01, 2023
Released on J-STAGE: March 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialThe importance of permeability as well as solubility of the drug has been recognized in improving the solubility of poorly water-soluble drugs. This study investigated the impact of amorphous composites of indomethacin (IMC) and sulindac (SLD) on the membrane permeability of drugs. The IMC/SLD (1/1) formulation prepared by dry grinding was amorphous with a single glass transition temperature. The Fourier transform IR spectra and Raman spectra revealed formation of hydrogen bonds between the OH group of IMC and the carbonyl group of SLD. These results suggest that an amorphous composite was formed between IMC and SLD through hydrogen bonding. The amount of dissolved IMC and SLD from the amorphous composite of IMC/SLD (1/1) was higher than that of the untreated IMC or SLD in the dissolution test. The permeated amounts and permeation rates of both drugs were enhanced by increasing the solubility of the amorphous composite. Conversely, the apparent membrane permeability coefficients (Papp) were almost same for untreated drugs and amorphous composites. In the case of hydroxypropyl-β-cyclodextrin and sodium dodecyl sulfate, Papp of the drugs decreased with the addition of these compounds, although the drug solubility was enhanced by the solubilization effect. This study revealed that an amorphous composite formed through hydrogen bonding is an attractive pharmaceutical way to enhance the permeated amount and permeation rate without changing the Papp of both the drugs.
View full abstractDownload PDF (1366K) Full view HTML
- |<
- <
- 1
- >
- >|