
- |<
- <
- 1
- >
- >|
-
Tetsuhiro Nemoto2023Volume 71Issue 2 Pages 78
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
JOURNAL FREE ACCESS FULL-TEXT HTMLDownload PDF (193K) Full view HTML
-
Shunta Sato, Wataru Sasaki, Tomoyuki Sekino, Tatsuhiko Yoshino, Masahi ...2023Volume 71Issue 2 Pages 79-82
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialMetallaphotoredox-catalyzed allylation represents an emerging synthetic methodology that enables allylic substitution using nucleophilic radical species. The C–H allylation of N-aryl tetrahydroisoquinolines is an innovative example in this area and allows access to synthetically useful precursors for the further derivatization of tetrahydroisoquinolines. However, previous methods have required the use of noble metals, which has hampered their application due to concerns over their sustainability. Here we report the C–H allylation of N-aryl tetrahydroisoquinolines using a cobalt/organophotoredox dual catalyst system. Based on precedent, control experiments and controlled irradiation experiments, a mechanism for the cobalt/photoredox-catalyzed allylation that involves a π-allyl cobalt complex is proposed.
View full abstractDownload PDF (599K) Full view HTML
-
Masafumi Ueda, Ayano Ichimonji, Miku Nakayama, Sachiko Ito, Norihiko T ...2023Volume 71Issue 2 Pages 83-92
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialDirect oxidation of the C(sp3)–H bond of β-(alkoxy)imino carbonyl compounds using copper acetate and molecular oxygen has been established. The protocol features a broad substrate scope and generates 1-imino-2,3-dicarbonyls in good to excellent yields.
View full abstractDownload PDF (762K) Full view HTML -
Yuichi Kuboki, Shohei Ohno, Makoto Sako, Kenichi Murai, Mitsuhiro Aris ...2023Volume 71Issue 2 Pages 93-100
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
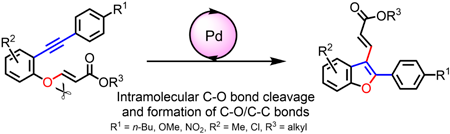
Supplementary materialMigratory cycloisomerization using transition metal catalyst is useful for synthesizing substituted heterocyclic compounds. We achieved palladium-catalyzed migratory cycloisomerization of 3-o-alkynylphenoxy acrylic acid ester derivatives to give 2,3-disubstituted benzofurans. Although there are several reports of benzofuran synthesis with palladium-catalyzed migratory cycloisomerization, migratory groups are limited to allyl and propargyl groups. This report is the first example of benzofuran synthesis with palladium-catalyzed cycloisomerization of C(sp2)-O bond cleavage.
View full abstractDownload PDF (705K) Full view HTML -
Lu Lin, Shunsuke Kataoka, Kiichi Hirayama, Ryozo Shibuya, Kenji Watana ...2023Volume 71Issue 2 Pages 101-106
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialCatalytic control of chemoselectivity is crucial in the synthesis of highly functionalized compounds. Although there are reports of efficient chemoselective reactions of alcohols and amines as nucleophiles, there are no reports of the chemoselective activation of alcohols and amines as electrophiles. In this study, highly O- and N-selective electrophilic activation of allylic alcohols and amines was achieved in Pd-catalyzed direct allylic alkylation. Allylamines were inherently more reactive than allylic alcohols (N-selectivity). On the other hand, the addition of catalytic amounts of 9-phenanthreneboronic acid preferentially activated allylic alcohols over allylamines (O-selectivity). Density functional theory (DFT) calculations suggested that the N-selectivity is due to the selective activation of allylic amines with ammonium cations, and boronate formation accelerates the activation of allylic alcohols.
View full abstractDownload PDF (823K) Full view HTML -
Tsukasa Furuya, Kotaro Ikeda, Shingo Harada, Tetsuhiro Nemoto2023Volume 71Issue 2 Pages 107-110
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialC–H insertion and amide insertion reactions using metal-carbene species provide a powerful synthetic method for direct functionalization of kinetically inert or thermodynamically stable chemical bonds. Our group previously developed an amide insertion reaction using a rhodium-dimer complex, constructing an array of nitrogen-bridged heterocycles. Another research group reported C–H insertion reactions using structurally related substrates and rhodium catalysts. Detailed mechanistic studies were not provided, however, and therefore, the origin of the chemoselectivity was ambiguous. Here we describe our theoretical investigation of the chemoselectivity between the amide insertion reaction and C–H functionalization. An energy gap of the identified transition states in the reaction coordinates could support the reported experimental results and the observed chemoselectivity. Moreover, frontier molecular orbital analysis revealed that functionalities adjacent to the metal-carbene species could affect orbital populations and their energy levels, resulting in the construction of a completely distinctive ring system.
View full abstractDownload PDF (3681K) Full view HTML
-
Qihui Xu, Yifan Wu, Hiroki Saito, Yuki Ofuchi, Haruna Setoyama, Takayu ...2023Volume 71Issue 2 Pages 111-119
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLFamotidine (FMT) is a competitive histamine-2 (H2) receptor antagonist that inhibits gastric acid secretion for the treatment of Gastroesophageal reflux disease. To study the promoting effect and mechanism of terpenes, including l-menthol, borneol, and geraniol, as chemical enhancers, FMT was used as a model drug. Attenuated total reflectance-Fourier transform IR spectroscopy (ATR-FTIR) and differential scanning calorimetry (DSC) were used to explore the effects of terpenes on the skin. Hairless mouse skin was mounted on Franz-type diffusion cell, and skin permeation experiment of FMT hydrogel was carried out. The results suggested that the thermodynamic activity influenced the permeability of the drug, and the main mechanism of terpenes to enhance skin permeation of the drug was based on increasing the fluidity of the intercellular lipids. Moreover, it was revealed that l-menthol simultaneously relaxed the packing structure and lamellar structure, whereas geraniol had a great influence on the lamellar structure only. Collectively, all terpenes had a promoting effect on skin permeation of FMT, indicating their potential as chemical enhancers to change the microstructure of stratum corneum and improve the permeation of FMT through the skin, and it has great potential to be used in transdermal formulations of FMT.
View full abstractDownload PDF (1618K) Full view HTML -
 Qi Zhang, Peizheng Yan, Pan Zhao, Dongsheng Zhao, Heran Cao, Jing Lu, ...2023Volume 71Issue 2 Pages 120-128
Qi Zhang, Peizheng Yan, Pan Zhao, Dongsheng Zhao, Heran Cao, Jing Lu, ...2023Volume 71Issue 2 Pages 120-128
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
Advance online publication: November 26, 2022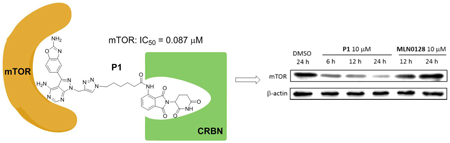 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialMechanistic target of rapamycin (mTOR) is an effective anti-tumor drug target. Several mTOR kinase inhibitors have entered clinical research, but there are still challenges of potential toxicity. As a new type of targeted drug, proteolysis targeting chimeras (PROTACs) have features of low dosage and low toxicity. However, this approach has been rarely reported to involve mTOR degradation. In this study, the mTOR kinase inhibitor MLN0128 was used as the ligand to the protein of interest and conjugated with pomalidomide by diverse intermediate linkage chains. Several potential small molecule PROTACs for the degradation of mTOR were designed and synthesized. PROTAC compounds exhibited mTOR inhibitory activity and suppressed MCF-7 cell proliferation. The representative compound P1 could inhibit the expression of mTOR downstream proteins and the growth of cancer cells by inducing autophagy but not affecting the cell cycle and not inducing apoptosis.
View full abstractEditor's pickIn this study, several novel PROTACs for the degradation of mTOR were designed based on MLN0128 (mTOR-binding ligand) and pomalidomide (E3 ligase CRBN ligand). PROTAC compounds exhibited mTOR inhibitory activity and suppressed MCF-7 cell proliferation. The representative compound P1 could degrade mTOR and reduce the expression of the mTOR downstream protein p-S6 (Ser240/244) and p-AKT (Ser473). Further studies showed that this compound could inhibit cancer cell growth by inducing autophagy, but it did not affect the cell cycle and apoptosis. This is the first mTOR PROTAC reported and these findings provide new insights in the study of mTOR inhibitors.
Download PDF (5164K) Full view HTML -
Yujin Wang, Na Dong, Yuan Zhou, Hongyan Li, Gangxin Qin, Hui Li, Qiaoq ...2023Volume 71Issue 2 Pages 129-133
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
Advance online publication: December 02, 2022 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLThis work aims to investigate the effects and mechanism of emodin in treating diabetic gastroenteropathy and colonic dysmotility in STZ + HS/HF diet induced diabetic gastroenteropathy rats. Diabetic colonic dysmotility model was established by high-fat/high-glucose (HS/HF) feeding combined with streptozotocin (STZ). Emodin was divided into high, medium and low dose groups. After eight weeks of intervention, fasting blood glucose (FBG) and body weight were measured. Gastrointestinal transmission time was evaluated. Serum vasoactive intestinal peptide (VIP) and substance P (SP) were detected. Colonic protein expression of selective autophagy adaptor proteins p62 and beclin1 were detected by immunohistochemistry. Colonic protein expression of beclin1, autophagy related gene 5 (Atg5), C-kit and p62 were detected by Western blot. After treating with emodin, gastrointestinal transmission rate was improved. The expression of serum SP was increased and serum VIP was decreased. Colonic c-kit and p62 were up-regulated. The expressions of beclin1 and Atg5 were down-regulated. Emodin can improve colonic dysmotility and promote the recovery of colonic motility and intestinal defecation in diabetic rats. Its mechanism may involved with up-regulating the expression of C-kit and P62, down-regulating the expression of Beclin1 and Atg5 in colon, which are associated with colon over-autophagy of Cajal interstitial cell (ICC).
View full abstractDownload PDF (3188K) Full view HTML -
Takeshi Goromaru, Kai Fujita, Misaki Mizumoto, Takashi Ishizu2023Volume 71Issue 2 Pages 134-139
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialA mixture of risperidone and eight major tea catechins: (−)-epicatechin-3-O-gallate (ECg), (−)-epigallocatechin-3-O-gallate (EGCg), (−)-catechin-3-O-gallate (Cg), (−)-gallocatechin-3-O-gallate (GCg), (−)-epicatechin (EC), (−)-epigallocatechin (EGC), (+)-catechin (CA), or (+)-gallocatechin (GC), in tartaric acid buffer (pH 3.0) afforded a precipitate. Amounts of risperidone and these catechins in the precipitate were measured by quantitative 1H-NMR (qNMR). About half or more of risperidone used was precipitated by gallated catechins ECg, EGCg, Cg, and GCg; on the other hand, it was precipitated little by non-gallated catechins EC, EGC, CA, and GC. Furthermore, risperidone was precipitated more by 2,3-trans gallated catechins Cg and GCg than 2,3-cis gallated catechins ECg and EGCg. Regarding the amount of tea catechins in the precipitate obtained by a mixture of risperidone and Catechin Mixture, the amounts of 2,3-cis gallated catechins EGCg and ECg were much larger than those of the other green tea catechins GCg, EC, EGC, CA, and GC. It was considered that risperidone was mainly precipitated by EGCg and ECg in Catechin Mixture. Therefore, it can be concluded that when patients take risperidone with catechin-rich beverages, the efficacy of risperidone reduces, mainly because the 2,3-cis gallated catechins EGCg and ECg form complexes with risperidone and precipitate.
View full abstractDownload PDF (2307K) Full view HTML -
Mingguang Zhang, Yang Yang, Yunyun Wang, Jia Wang, Hongyan Wu, Yongqia ...2023Volume 71Issue 2 Pages 140-147
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
Advance online publication: December 15, 2022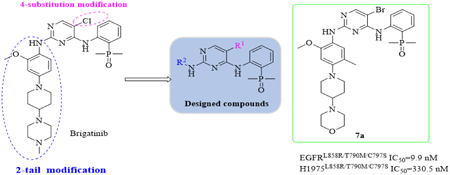 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialEpidermal growth factor receptor (EGFR) C797S mutation leads to Osimertinib drug resistance by disturbing the covalent biding of Michael acceptor group to the Cys797 residue in the ATP biding cleft. In this manuscript, a class of 2-amine-4-oxyphosaniline pyrimidine derivatives were designed, synthesized and evaluated as new noncovalent reversible EGFR inhibitors against L858R/T790M/C797S (CTL) triple mutant. The kinases inhibitiory activity evaluation showed that four compounds exhibited significant inhibitory activities against CTL (IC50 < 30 nM). In particularly, the most promising compound 7a showed excellent enzymatic inhibitory activity against CTL with IC50 value of 9.9 nM, which was more potent than control compound Osimertinib. Moreover, cell proliferation assays indicated that 7a effectively inhibited H1975-EGFR L858R/T790M/C797S with IC50 value of 0.33 µM. Furthermore, compound 7a displayed good metabolic stabilities in human, rat and mouse liver microsomes, and the putative biding mode of compound 7a with ATP was revealed by molecular docking study. These findings strongly indicated that compound 7a was a promising L858R/T790M/C797S mutant EGFR inhibitor.
View full abstractEditor's pickNon-small cell lung cancer (NSCLC) is the most common type of lung cancers. However, drug resistance via an acquired triple EGFR mutation were inevitably observed after treatment with current inhibitors. So far, there are no effective therapeutic strategies to overcome the L858R/T790M/C797S triple mutation. In this paper, a class of 2-amine-4-oxyphosaniline pyrimidine derivatives were developed to overcome L858R/T790M/C797S (CTL) triple mutant drug resistance, and a candidate compound was discovered and showed good activity against L858R/T790M/C797S triple mutant in vitro.
Download PDF (1548K) Full view HTML -
Rio Uno, Kyoko Ohkawa, Honami Kojima, Tamami Haraguchi, Minoru Ozeki, ...2023Volume 71Issue 2 Pages 148-153
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialThis study aimed to evaluate the bitterness of famotidine (FAM) combined with each of three non-steroidal anti-inflammatory drugs (NSAIDs): ibuprofen (IBU), flurbiprofen (FLU), and naproxen (NAP), which have potential as fixed-dose combination (FDC) drugs. We evaluated the bitterness of FAM and each NSAID by taste sensor AN0 and C00, respectively. FAM showed high sensor output representing sensitivity to bitterness, whereas three NSAIDs did not show large sensor output, suggesting that the bitterness intensities of three NSAIDs were lower than that of FAM. The bitterness of FAM on sensor AN0 was suppressed in a concentration-dependent manner when mixed with IBU, FLU, or NAP. Among three NSAIDs, IBU most effectively inhibited bitterness on sensor output, and the gustatory sensation test confirmed that adding IBU to FAM reduced the bitterness of FAM in a concentration-dependent manner. MarvinSketch confirmed that the drugs were mostly present in an ionic solution when FAM was mixed with NSAIDs. The 1H-NMR spectroscopy analysis also revealed the presence of electrostatic interactions between FAM and NSAIDs, suggesting that the electrostatic interaction between FAM and NSAIDs might inhibit the adsorption of FAM on the bitter taste sensor membrane, thereby masking the bitter taste.
View full abstractDownload PDF (1009K) Full view HTML -
Marie Kurihara, Vera Thiel, Hirona Takahashi, Keiichi Kojima, David M. ...2023Volume 71Issue 2 Pages 154-164
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
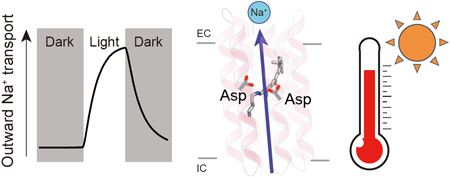
Supplementary materialRhodopsins are transmembrane proteins with retinal chromophores that are involved in photo-energy conversion and photo-signal transduction in diverse organisms. In this study, we newly identified and characterized a rhodopsin from a thermophilic bacterium, Bellilinea sp. Recombinant Escherichia coli cells expressing the rhodopsin showed light-induced alkalization of the medium only in the presence of sodium ions (Na+), and the alkalization signal was enhanced by addition of a protonophore, indicating an outward Na+ pump function across the cellular membrane. Thus, we named the protein Bellilinea Na+-pumping rhodopsin, BeNaR. Of note, its Na+-pumping activity is significantly greater than that of the known Na+-pumping rhodopsin, KR2. We further characterized its photochemical properties as follows: (i) Visible spectroscopy and HPLC revealed that BeNaR has an absorption maximum at 524 nm with predominantly (>96%) the all-trans retinal conformer. (ii) Time-dependent thermal denaturation experiments revealed that BeNaR showed high thermal stability. (iii) The time-resolved flash-photolysis in the nanosecond to millisecond time domains revealed the presence of four kinetically distinctive photointermediates, K, L, M and O. (iv) Mutational analysis revealed that Asp101, which acts as a counterion, and Asp230 around the retinal were essential for the Na+-pumping activity. From the results, we propose a model for the outward Na+-pumping mechanism of BeNaR. The efficient Na+-pumping activity of BeNaR and its high stability make it a useful model both for ion transporters and optogenetics tools.
View full abstractDownload PDF (4369K) Full view HTML -
Yoshihisa Yamamoto, Kosuke Ohgi, Yoshinori Onuki, Toshiro Fukami, Tats ...2023Volume 71Issue 2 Pages 165-174
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialIn the present study, we conducted a detailed evaluation of the effects of humidification on the quality of five types of commercial magnesium oxide (MgO) tablet formulations. When near-IR spectroscopy was performed, a peak derived from the first overtone of the stretching vibration of the hydroxyl group was observed at approximately 7200 cm−1 in a humidified MgO tablet formulation. To visually evaluate the effect of this humidification, a mapping image was created using microscopic IR spectroscopy. In the IR spectrum, a peak derived from the stretching vibration of the hydroxyl group appears at approximately 3700 cm−1, so we created a mapping image using the absorbance ratio of 3700 and 3400 cm−1 as an index. In the mapping image of humidified MgO tablet formulations, many areas had a higher absorbance ratio than the dried tablet formulations. From these results, it is qualitatively confirmed that the MgO was changed to magnesium hydroxide (Mg(OH)2) by humidification. Although these results were observed in the four types of MgO tablet formulations, only one type of tablet formulation was less affected by humidification. In addition, although most tablet formulations tended to prolong disintegration time due to humidification, there was almost no effect of humidification on the disintegration time in one type of tablet formulation, which had little change in the above evaluation. Thus, in most commercial MgO tablet formulations, humidification prolongs the disintegration time, and Mg(OH)2 significantly contributes to this factor.
View full abstractEditor's pickAuthors conducted a detailed evaluation of the effects of humidification on the quality of five types of commercial magnesium oxide (MgO) tablet formulations by near-infrared spectroscopy, microscopic infrared spectroscopy and thermogravimetry. From these analysis results, it is qualitatively confirmed that the MgO was changed to magnesium hydroxide by humidification. In addition, most tablet formulations tended to prolong disintegration time due to humidification. Thus, in most commercial MgO tablet formulations, it is suggested magnesium hydroxide significantly contributes to prolongation of disintegration time by humidification. The results obtained in this study will provide useful information regarding the handling of MgO tablets in medical sites.
Download PDF (4843K) Full view HTML -
Keisuke Kinoshita, Miyuki Yamaguchi, Hirohisa Sasou, Hideyuki Konishi, ...2023Volume 71Issue 2 Pages 175-182
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
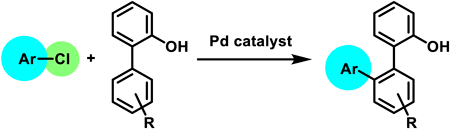 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialPalladium-catalyzed, hydroxy-group-directed C–H arylation of [1,1′-biphenyl]-2-ols with chloroarenes was performed. The reaction showed a broad substrate scope and was successfully applied to pharmaceuticals containing a chloro group. Using 2-heteroarylphenols instead of [1,1′-biphenyl]-2-ols also yielded the desired products. The arylated product was further transformed into a triphenylene derivative.
View full abstractEditor's pickHydroxy-directed, Pd-catalyzed C‒H arylation of [1,1'-biphenyl]-2-ol with haloarenes, developed by Miura et al., is a useful method to synthesize ortho-teraryls. However, only bromo- and iodoarenes were used as arylating agents. In this paper, the authors report that chloroarenes including chloro-containing pharmaceuticals were successfully used as haloarenes for the reactions under optimized reaction conditions. In addition, it was revealed that substituted [1,1'-biphenyl]-2-ols and 2-heteroarylphenols instead of [1,1'-biphenyl]-2-ol were also usable for the reactions. Transformation of the ortho-teraryl product into a triphenylene derivative is also presented.
Download PDF (934K) Full view HTML
-
Thanh Hao Huynh, Zhi-Hong Wen, Jui-Hsin Su, Zhi-Kang Yao, Li-Guo Zheng ...2023Volume 71Issue 2 Pages 183-187
Published: February 01, 2023
Released on J-STAGE: February 01, 2023
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialA formerly unpublicized briarane diterpenoid, briastecholide M (1), and its established analogue, brianodin B (2), were purified from Briareum stechei, an octocoral collected from Okinawan waters. Using spectroscopic methods, the structure of 1 was established. Functional study showed that 1 can reducing the release of inducible nitric oxide synthase (iNOS) but enhancing cyclooxygenase-2 (COX-2) protein expression.
View full abstractDownload PDF (771K) Full view HTML
- |<
- <
- 1
- >
- >|