
- |<
- <
- 1
- >
- >|
-
 Yusuke Sato2021Volume 69Issue 12 Pages 1141-1159
Yusuke Sato2021Volume 69Issue 12 Pages 1141-1159
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLConsiderable efforts have been made on the development of lipid nanoparticles (LNPs) for delivering of nucleic acids in LNP-based medicines, including a first-ever short interfering RNA (siRNA) medicine, Onpattro, and the mRNA vaccines against the coronavirus disease 2019 (COVID-19), which have been approved and are currently in use worldwide. The successful rational design of ionizable cationic lipids was a major breakthrough that dramatically increased delivery efficiency in this field. The LNPs would be expected to be useful as a platform technology for the delivery of various therapeutic modalities for genome editing and even for undiscovered therapeutic mechanisms. In this review, the current progress of my research, including the molecular design of pH-sensitive cationic lipids, their applications for various tissues and cell types, and for delivering various macromolecules, including siRNA, antisense oligonucleotide, mRNA, and the clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) system will be described. Mechanistic studies regarding relationships between the physicochemical properties of LNPs, drug delivery, and biosafety are also summarized. Furthermore, current issues that need to be addressed for next generation drug delivery systems are discussed.
View full abstractEditor's pickLipid nanoparticles (LNPs) are able to deliver various therapeutic macromolecules, including short interference RNAs, mRNAs, and the clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) ribonucleoproteins, into the cytosol of the target cells through endocytosis followed by membrane fusion-mediated endosomal escape to induce gene silencing, gene expression, and gene editing, respectively. Rational molecular design of pH-sensitive cationic lipid was a major breakthrough that dramatically increased delivery efficiency in this field. The LNPs would be expected to be useful as a platform technology for the delivery of various therapeutic modalities for genome editing and even for undiscovered therapeutic mechanisms.
Download PDF (7966K) Full view HTML -
Hidetoshi Noda2021Volume 69Issue 12 Pages 1160-1169
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
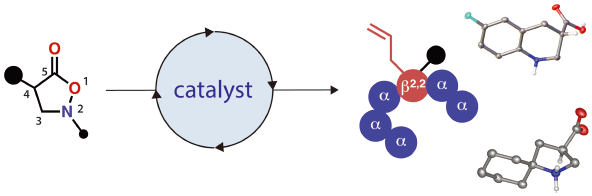
JOURNAL FREE ACCESS FULL-TEXT HTMLIsoxazolidin-5-ones have been regarded as β-amino acid surrogates owing to their labile N–O bond. While many efforts have been devoted to the catalytic enantioselective synthesis of the core of this heterocycle, its further transformation has been less explored, especially in the context of catalysis. This review summarizes the author’s research on the development of catalytic reactions using isoxazolidin-5-ones as substrates. Asymmetric catalysis has proven effective for C–C bond formation at the carbonyl α-carbon. Catalytic asymmetric allylation and direct Mannich-type reactions have been developed. Further, the resulting products have been readily converted into the corresponding quaternary β2,2-amino acids. Moreover, isoxazolidin-5-ones have been identified as alkyl nitrene precursors in the presence of a suitable metal catalyst. The generated metallonitrene undergoes either the electrophilic amination of the aromatic ring or aliphatic C–H insertion, affording a series of cyclic β-amino acids. A remarkable difference in chemoselectivity between rhodium and copper alkyl nitrenes has also been demonstrated, highlighting the unique nature of the underexplored reactive intermediates. The various linear and cyclic β-amino acids obtained through the study are likely to find great utility in a broad range of chemical sciences.
View full abstractEditor's pickCatalysts have provided new modes of activation that enable the transformation of otherwise inert molecules. Owing to the labile nature of the N–O bond under reductive conditions, isoxazolidin-5-ones have been used as useful b-amino acid surrogates. In the last decade, this century-old heterocycle has experienced a renaissance upon the merger with catalysis, allowing for the synthesis of structurally diversified molecules otherwise difficult to obtain. This review article highlights the author’s research in this emerging field, focusing on the catalytic asymmetric a-functionalization of – and the alkyl nitrene formation from – the heterocycle.
Download PDF (1813K) Full view HTML -
Shingo Harada2021Volume 69Issue 12 Pages 1170-1178
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
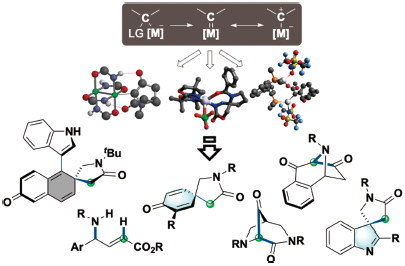
JOURNAL FREE ACCESS FULL-TEXT HTMLThe ability to control the reactions of highly active chemical species to enable straightforward synthesis of valuable compounds such as bioactive natural products and pharmaceuticals is a continuing challenge in synthetic organic chemistry. This review describes the development of a methodology using reactive metal-carbene species and its synthetic application in our laboratory. First, regioselective synthesis of γ-amino acid equivalents to take advantage of their metal-dependent reactivities and the mechanistic rationale are presented. Chemoselective and enantioselective dearomatization reactions of several arenes with silver-carbene are also discussed. In the second half of the review, we discuss a carbene-insertion reaction into an amide and urea C–N bond for the assembly of nitrogen-bridged cyclic molecules.
View full abstractEditor's pickThe author focuses on expanding metal-carbene chemistry via developing novel reactions, mechanistic analysis based on computational predictions, and synthetic demonstrations for pharmaceutically essential molecules. The findings include that silver-carbenes possess unusual chemical properties, leading to unique chemoselectivities and regioselectivities with stereocontrol. By utilizing the exceptional reactivities, the enantioselective reactions involving silver-carbenes were achieved in studies regarding arene dearomatization. A novel type of insertion reactions was also developed using a designed Rh2(NHCOtBu)4 catalyst, assembling a series of nitrogen-bridged heterocycles. The author believes that the obtained mechanistic profiles could serve as bases for designing novel reactions and advanced catalysis for pharmaceuticals.
Download PDF (2050K) Full view HTML
-
Taichi Kamo, Keiichi Kuroda, Shota Kondo, Usaki Hayashi, Satoshi Fudo, ...2021Volume 69Issue 12 Pages 1179-1183
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialMetallo-β-lactamases (MBLs) are significant threats to humans because they deteriorate many kinds of β-lactam antibiotics and are key enzymes responsible for multi-drug resistance of bacterial pathogens. As a result of in vitro screening, two compounds were identified as potent inhibitors of two kinds of MBLs: imipenemase (IMP-1) and New Delhi metallo-β-lactamase (NDM-1). The binding structure of one of the identified compounds was clarified by an X-ray crystal analysis in complex with IMP-1, in which two possible binding poses were observed. Molecular dynamics (MD) simulations were performed by building two calculation models from the respective binding poses. The compound was stably bound to the catalytic site during the simulation in one pose. The binding model between NDM-1 and the compound was constructed for MD simulation. Calculation results for NDM-1 were similar to those of IMP-1. The simulation suggested that the binding of the identified inhibitory compound was also durable in the catalytic site of NDM-1. The compound will be a sound basis for the development of the inhibitors for MBLs.
View full abstractDownload PDF (3155K) Full view HTML -
Daisuke Mizunaga, Mika Koseki, Naoki Kamemoto, Satoru Watano2021Volume 69Issue 12 Pages 1184-1194
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTMLA quantitative evaluation method for determining the effect of tableting speed on the compression properties of pharmaceutical powders was investigated in this study. Cilostazol and ibuprofen were used as active pharmaceutical ingredients (APIs) and mixed with lactose monohydrate and microcrystalline cellulose. Viscoelasticity was examined to evaluate the raw material, and stress relaxation tests were conducted to determine the apparent viscosity and elasticity coefficients of the placebo and two APIs. Tablets were prepared using a compaction simulator and a rotary tablet press at the tableting speeds ranging from laboratory to commercial. The in-die or out-of-die strain rate sensitivity (SRS) indices were determined as a measure of the compressibility and compactibility. The results showed that the sensitivity of the out-of-die SRS was higher than that of the in-die SRS. The out-of-die SRS of ibuprofen 20% powder, which showed high elasticity and low viscosity, was 13.3–47.9%, whereas that of the placebo and cilostazol 20% (w/w) powder was <7.5%. A peripheral speed difference of more than 300 mm/s during the out-of-die SRS was sensitive enough to detect the capping tendency. Cilostazol, which has lower elasticity and higher viscosity than ibuprofen, was tested using powder mixtures with the API concentrations of 5–40%; the compressibility SRS was <5% for all API concentrations. In contrast, the compressibility SRS of ibuprofen increased from 4.8 to 81% depending on the API concentration. Using the compressibility SRS as an index, it was possible to extract the tableting speed-dependent compressibility characteristics of API from the powder mixtures containing API.
View full abstractDownload PDF (1233K) Full view HTML -
Yutaka Suto, Tatiana Ascencio, Tomoya Nobuta, Noriyuki Yamagiwa, Yoko ...2021Volume 69Issue 12 Pages 1195-1199
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
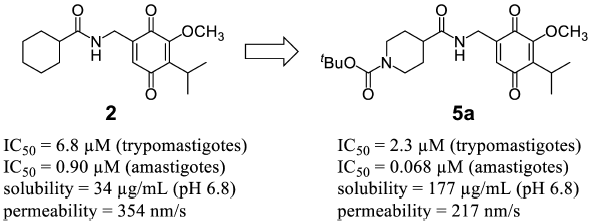
Supplementary materialA series of quinone derivatives with a variety of side chains were synthesized. These synthetic quinone compounds were evaluated for in vitro antitrypanosomal activity against trypomastigotes and amastigotes of Trypanosoma cruzi, the causative agent of Chagas disease. Measurement of solubility of quinones and their ability to permeate cell membranes were assessed to address their possible use as oral drugs. Some synthesized compounds exhibited potent antitrypanosomal activity. However, most compounds with a promising activity showed poor solubility that did not seem suitable for oral usage. Meanwhile, compound 5a, an N-tert-butoxycarbonylpiperidine derivative, exhibited good antitrypanosomal activity, ability to permeate membranes, and good solubility.
View full abstractEditor's pickChagas disease is a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi and an important health problem in rural areas of Latin American countries. Safe drugs with high clinical efficacy have been urgently needed. Authors synthesized quinone compounds and evaluated their in vitro antitrypanosomal activity against trypomastigotes and amastigotes of Trypanosoma cruzi. In addition, solubility and membrane permeability were measured to confirm whether compounds with promising antiprotozoal activity had physicochemical properties compatible with oral administration. Among the synthesized compounds, 5a showed adequate antiprotozoal activity, solubility, and high membrane permeability.
Download PDF (446K) Full view HTML -
Kwihwan Park, Jing Jiang, Tsuyoshi Yamada, Hironao Sajiki2021Volume 69Issue 12 Pages 1200-1205
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialA protocol for the ruthenium-on-carbon (Ru/C)-catalyzed solvent-free oxidation of alcohols, which proceeds efficiently under solid–solid (liquid)–gas conditions, was developed. Various primary and secondary alcohols were transformed to corresponding aldehydes and ketones in moderate to excellent isolated yields by simply stirring in the presence of 10% Ru/C under air or oxygen conditions. The solvent-free oxidation reactions proceeded efficiently regardless of the solid or liquid state of the substrates and reagents and could be applied to gram-scale synthesis without loss of the reaction efficiency. Furthermore, the catalytic activity of Ru/C was maintained after five reuse cycles.
View full abstractDownload PDF (770K) Full view HTML
-
Katsuhiko Sato, Kazuhiro Watanabe, Kyoko Sugiyama, Sachiko Komatsu, Ts ...2021Volume 69Issue 12 Pages 1206-1208
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
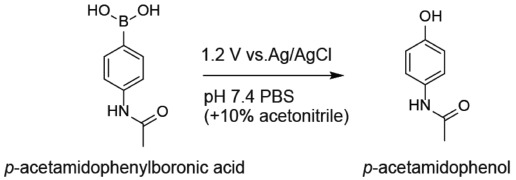
Supplementary materialHerein, it is reported that p-acetamidophenylboronic acid can be electrolytic cleavage of the carbon–boron bond to p-acetamidophenol at an electric potential of 1.2 V vs. Ag/AgCl in 100 mM phosphate buffer of pH 7.4 (containing 10% acetonirile). The electrochemical reaction was investigated by HPLC, LC with tandem mass spectrometry, and cyclic voltammetry. This electrochemical reaction could be useful in the development of electrical controlled drug delivery systems under neutral pH conditions.
View full abstractDownload PDF (448K) Full view HTML -
Shota Machida, Maho Sugaya, Hiroaki Saito, Taketo Uchiyama2021Volume 69Issue 12 Pages 1209-1212
Published: December 01, 2021
Released on J-STAGE: December 01, 2021
 JOURNAL FREE ACCESS FULL-TEXT HTML
JOURNAL FREE ACCESS FULL-TEXT HTML
Supplementary materialGallotannins are phenolic natural products containing galloyl moieties connected to polyhydric alcohol cores, e.g., D-glucose. Some gallotannins are reported to have antidiabetic properties, such as α-glucosidase inhibitory activity. In this study, fourteen unnatural gallotannin derivatives with 1,5-anhydroalditol and inositol as the cyclic polyol cores were synthesized to investigate how their structures affected antioxidative and α-glucosidase inhibitory activities. Tannic acid demonstrated the most potent antioxidative activity (EC50 = 2.84 μM), with potency increasing proportionally to the number of galloyl moieties. Synthetic inositol derivatives outperformed 1,5-anhydroalditol derivatives in rat α-glucosidase inhibitory activity. Pentagalloyl glucose, a natural compound, demonstrated the highest activity (IC50 = 0.336 μM).
View full abstractDownload PDF (553K) Full view HTML
- |<
- <
- 1
- >
- >|